GenomeScope 是2017年发表在 bioinformatic 的一个工具,这个工具的目的就是处理一些高复杂度的基因组,比如说高杂合度或者基因组非常大的物种。GenomeScope只能预测二倍体基因组,GenomeScope 2.0可以预测多倍体物种。
安装
$ git clone https://github.com/tbenavi1/genomescope2.0.git
$ cd genomescope2.0/
$ Rscript install.R
在软件的安装目录下,genomescopre.R文件是核心的运行脚本,用法如下
$ Rscript genomescope.R \
-i histogram_file \
-o output_dir \
-k k-mer_length
可选参数:
-i input histogram_file (from KMC or jellyfish) ,如jellyfish软件产生的kmer频数分布数据
-o output_dir
-k kmer length used to calculate kmer spectra [default 21] ;
必选参数:
-p PLOIDY, --ploidy PLOIDY ploidy (1, 2, 3, 4, 5, or 6) for model to use [default 2] ;
-m MAX_KMERCOV, --max_kmercov MAX_KMERCOV optional maximum kmer coverage threshold (kmers with coverage greater than max_kmercov are ignored by the model) ;
-n NAME_PREFIX, --name_prefix NAME_PREFIX optional name_prefix for output files ;
-l LAMBDA, --lambda LAMBDA, --kcov LAMBDA, --kmercov LAMBDA optional initial kmercov estimate for model to use ;
示例:
/path/R/3.1.1/bin/Rscript /path/genomescope2.0/genomescope.R
-i /home/liuliu/test/Survey//kmer_21/21mer.out.hist \
-o /home/liuliu/test/Survey/kmer_21/output_p3 \
-k 21 \
-p 3 \
-n 21.p3 \
在运行过程中,终端会输出如下信息
GenomeScope analyzing /home/liuliu/test/Survey/kmer_21/21mer.out.hist p=3 k=21 outdir=/home/liuliu/test/Survey//kmer_21/output_p3
aaa:98.4% aab:1.51% abc:0.14%
Model converged het:0.0165 kcov:18.4 err:0.00279 model fit:0.541 len:376278986
het表示杂合度,为1.65%;len表示基因组大小,为376M左右。
输出目录output_p3文件列表如下
|-- 21.p3_linear_plot.png
|-- 21.p3_log_plot.png
|-- 21.p3_model.txt
|-- 21.p3_progress.txt
|-- 21.p3_summary.txt
|-- 21.p3_transformed_linear_plot.png
`-- 21.p3_transformed_log_plot.png
0 directories, 7 files
通常关注summary.txt, transformed_linear_plot.png这2个文件。
1. summary.txt
内容如下:
$cat 21.p2_summary.txt
GenomeScope version 2.0
input file = /home/liuliu/test/Survey//kmer_21/21mer.out.hist
output directory = /home/liuliu/test/Survey/kmer_21/output_p3
p = 3
k = 21
name prefix = 21.p3
property min max
Homozygous (aaa) 98.3038% 98.4035%
Heterozygous (not aaa) 1.5965% 1.69625%
aab 1.47668% 1.53603%
abc 0.119818% 0.160215%
Genome Haploid Length 375,321,002 bp 376,278,986 bp
Genome Repeat Length 124,068,954 bp 124,385,633 bp
Genome Unique Length 251,252,048 bp 251,893,353 bp
Model Fit 72.9102% 97.1951%
Read Error Rate 0.279346% 0.279346%
在该文件中,会给出杂合度,基因组大小,重复片段长度等详细信息。
结果分为三列:
- 第一列为统计指标;
- 第二列为对应指标预测的最小值;
- 第三列为对应指标预测的最大值
有疑问,可以对照模型进行检验。
2. transformed_linear_plot.png
Occasionally, it is difficult to determine whether a peak in the kmer spectrum is a major peak. For this reason, GenomeScope 2.0 analyzes a transformed k-mer spectrum, in which the heights of higher-order peaks are increased.
K-mer覆盖度-频数分布图如下:
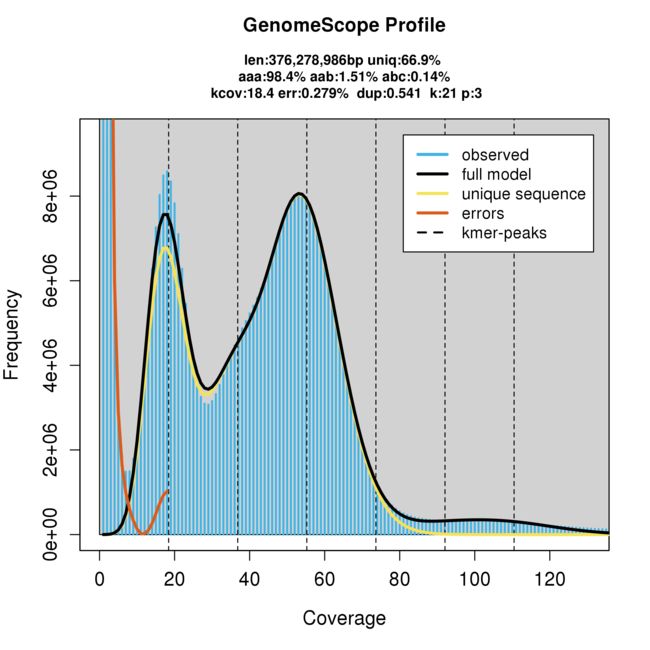
- 蓝色柱子是实际观测值;
- 黑色拟合线是除去被认为是错误的部分(橙红色拟合线部分)之后剩下的所有k-mer,这些被认为是可靠的kmer数据,只拿这部分数据来评估基因组的大小;
- 黄色拟合线被认为来自基因组非重复区域的K-mer分布;
- 橙红色拟合线部分对应着深度过低的kmer,这些kmer被认为是测序错误引入的;
- 垂直的黑色虚线为预测最低深度峰的整倍数覆盖度;
kcov指的是杂合峰的覆盖度。可以看到使用数据预测K-mer最低深度峰在18.4X处。一般情况下杂合度大于1%就会存在一个高于主峰的杂合峰。
基因组越大,杂合度也大,重复片段越大,该物种的组装难度就越大。
讨论:
基因组预测大小和参数 Max kmer coverage 密切相关。GenomeScope默认会过滤掉出现10,000次以上的kmers,避免细胞器基因组的影响,如果你觉得基因组小了,那么就把数值调整的大一点。
基因组survey介绍了如何通过jellyfish统计k-mer然后绘制k-mer分布图研究基因组的方法。
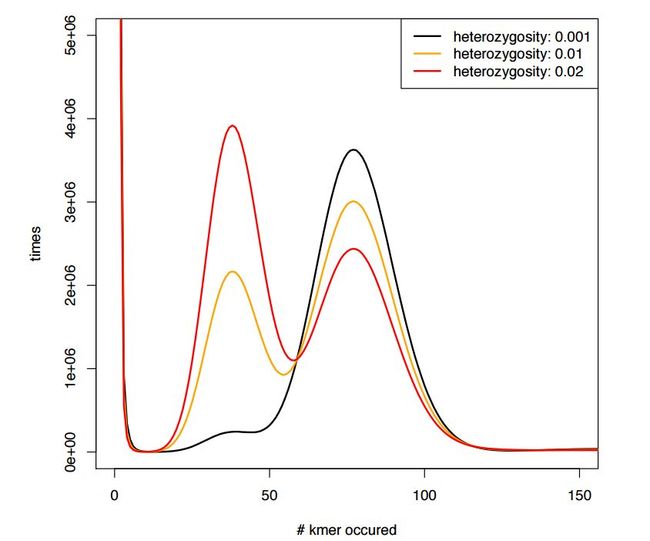
对于不同的基因组杂合度,kmer分布如下
https://github.com/tbenavi1/genomescope2.0