导读
举例展示R语言组学关联分析的方法。宏基因组数据以KO-样品丰度表为例。代谢组数据以metabolite-样品丰度表为例。基本方法是用R语言psych包corr.test函数进行两组数据的相关分析,结果经格式化后用pheatmap可视化得热图。
一、模拟输入
1. KO丰度表
ko_abun = as.data.frame(matrix(abs(round(rnorm(200, 100, 10))), 10, 20))
colnames(ko_abun) = paste("KO", 1:20, sep="_")
rownames(ko_abun) = paste("sample", 1:10, sep="_")
ko_abun

2. metabolite丰度表
metabo_abun = as.data.frame(matrix(abs(round(rnorm(200, 200, 10))), 10, 20))
colnames(metabo_abun) = paste("met", 1:20, sep="_")
rownames(metabo_abun) = paste("sample", 1:10, sep="_")
metabo_abun

二、相关分析函数
思路
- 参数一:other -> KO或其他组学丰度表
- 参数二:metabo -> 代谢物丰度表
- 参数三:route -> 输出目录【提前创建】
- corr.test进行两组数据相关分析
- 用stringr split将ko-metabolite结果列拆成两列
- 结果保留r_value p_value
library(psych)
library(stringr)
correlate = function(other, metabo, route)
{
# 读取方式:check.name=F, row.names=1, header=T
# 计算相关性:
#other = data
#metabo = env
#route="gut"
result=data.frame(print(corr.test(other, metabo, use="pairwise", method="spearman", adjust="fdr", alpha=.05, ci=TRUE, minlength=100), short=FALSE, digits=5))
# FDR矫正
result_raw=data.frame(print(corr.test(other, metabo, use="pairwise", method="spearman", adjust="none", alpha=.05, ci=TRUE, minlength=100), short=FALSE, digits=5))
# 原始P value
# 整理结果
pair=rownames(result) # 行名
result2=data.frame(pair, result[, c(2, 4)]) # 提取信息
# P值排序
# result3=data.frame(result2[order(result2[,"raw.p"], decreasing=F),])
# 格式化结果【将细菌代谢物拆成两列】
result4=data.frame(str_split_fixed(result2$pair, "-", 2), result2[, c(2, 3)], p_value=result_raw[, 4])
colnames(result4)=c("feature_1", "feature_2", "r_value", "fdr_p_value", "raw_p_value")
# 保存提取的结果
write.table(result4, file=paste(route, "Correlation_result.txt", sep="/"), sep="\t", row.names=F, quote=F)
}
三、相关性分析
dir.create("Result") # 创建结果目录
correlate(ko_abun, metabo_abun, "Result")
- 结果Correlation_result.txt如下,行数为20X20

四、pheatmap可视化函数
思路:
- 参数一:infile -> Correlation_result.txt相关性分析结果
- 参数二:route -> Route输出路径
- 用reshape2 dcast函数把feature_1 feature_2 p_value做成matrix,作为pheatmap输入文件
- 用reshape2 dcast函数把feature_1 feature_2 r_value做成matrix,作为pheatmap display
- 显著相关标记* 0.05>=p>0.01; ** 0.01>=p>0.001; *** 0.001>=p
library(reshape2)
library(pheatmap)
correlate_pheatmap = function(infile, route)
{
data=read.table(paste(route, infile, sep="/"), sep="\t", header=T)
data_r=dcast(data, feature_1 ~ feature_2, value.var="r_value")
data_p=dcast(data, feature_1 ~ feature_2, value.var="raw_p_value")
rownames(data_r)=data_r[,1]
data_r=data_r[,-1]
rownames(data_p)=data_p[,1]
data_p=data_p[,-1]
# 剔除不显著的行
del_row = c()
for(i in 1:length(data_p[, 1]))
{
if(all(data_p[i, ] > 0.05))
{
del_row = c(del_row, i)
}
}
# 剔除不显著的列
del_col = c()
for(j in 1:length(data_p[1, ]))
{
if(all(data_p[, j] > 0.05))
{
del_col = c(del_col, j)
}
}
# null值处理
if(is.null(del_row) && !(is.null(del_col)))
{
data_p = data_p[, -del_col]
data_r = data_r[, -del_col]
}else if(is.null(del_col) && !(is.null(del_row)))
{
data_p = data_p[-del_row,]
data_r = data_r[-del_row,]
}else if(is.null(del_row) && is.null(del_col))
{
print("delete none")
}else if(!(is.null(del_row)) && !(is.null(del_col)))
{
data_p = data_p[-del_row, -del_col]
data_r = data_r[-del_row, -del_col]
}
# data_p = data_p[-del_row, -del_col]
# data_r = data_r[-del_row, -del_col]
write.csv(data_p, file=paste(route, "data_p.csv", sep="/"))
write.csv(data_r, file=paste(route, "data_r.csv", sep="/"))
# 用"*"代替<=0.05的p值,用""代替>0.05的相对丰度
data_mark=data_p
for(i in 1:length(data_p[,1])){
for(j in 1:length(data_p[1,])){
#data_mark[i,j]=ifelse(data_p[i,j] <= 0.05, "*", "")
if(data_p[i,j] <= 0.001)
{
data_mark[i,j]="***"
}
else if(data_p[i,j] <= 0.01 && data_p[i,j] > 0.001)
{
data_mark[i,j]="**"
}
else if(data_p[i,j] <= 0.05 && data_p[i,j] > 0.01)
{
data_mark[i,j]="*"
}
else
{
data_mark[i,j]=""
}
}
}
write.csv(data_mark, file=paste(route, "data_mark.csv", sep="/"))
pheatmap(data_r, display_numbers=data_mark, cellwidth=20, cellheight=20, fontsize_number=18, filename=paste(route, "Correlation_result.pdf", sep="/"))
pheatmap(data_r, display_numbers=data_mark, cellwidth=20, cellheight=20, fontsize_number=18, filename=paste(route, "Correlation_result.png", sep="/"))
}
五、pheatmap绘制热图
correlate_pheatmap("Correlation_result.txt", "Result")
- 结果目录,新增结果图(一个png一个pdf):

- 打开pdf,如下:

- 随机模拟的数据,没有显著的不奇怪。如果是做项目一般两组数据的相关分析都可以得到一些显著相关的结果,如下:

两组数据完全相同的话:

六、绘图展示 |r| > 0.8的相关
1 函数
select_pheatmap = function(infile, route)
{
data=read.table(paste(route, infile, sep="/"), sep="\t", header=T)
data_r=dcast(data, feature_1 ~ feature_2, value.var="r_value")
data_p=dcast(data, feature_1 ~ feature_2, value.var="raw_p_value")
rownames(data_r)=data_r[,1]
data_r=data_r[,-1]
rownames(data_p)=data_p[,1]
data_p=data_p[,-1]
# 记下0.8 > r > -0.8 的行
del_row = c()
for(i in 1:length(data_p[, 1]))
{
if(all(data_r[i, ] < 0.8 & data_r[i, ] > -0.8))
{
del_row = c(del_row, i)
}
}
# 记下0.8 > r > -0.8 的列
del_col = c()
for(j in 1:length(data_p[1, ]))
{
if(all(data_r[, j] < 0.8 & data_r[, j] > -0.8))
{
del_col = c(del_col, j)
}
}
# null值处理
if(is.null(del_row) && !(is.null(del_col)))
{
data_p = data_p[, -del_col]
data_r = data_r[, -del_col]
}else if(is.null(del_col) && !(is.null(del_row)))
{
data_p = data_p[-del_row,]
data_r = data_r[-del_row,]
}else if(is.null(del_row) && is.null(del_col))
{
print("delete none")
}else if(!(is.null(del_row)) && !(is.null(del_col)))
{
data_p = data_p[-del_row, -del_col]
data_r = data_r[-del_row, -del_col]
}
# data_p = data_p[-del_row, -del_col]
# data_r = data_r[-del_row, -del_col]
write.csv(data_p, file=paste(route, "data_p.csv", sep="/"))
write.csv(data_r, file=paste(route, "data_r.csv", sep="/"))
# 用"*"代替<=0.05的p值,用""代替>0.05的相对丰度
data_mark=data_p
for(i in 1:length(data_p[,1])){
for(j in 1:length(data_p[1,])){
if(data_p[i,j] <= 0.001)
{
data_mark[i,j]="***"
}
else if(data_p[i,j] <= 0.01 && data_p[i,j] > 0.001)
{
data_mark[i,j]="**"
}
else if(data_p[i,j] <= 0.05 && data_p[i,j] > 0.01)
{
data_mark[i,j]="*"
}
else
{
data_mark[i,j]=""
}
}
}
# r值mark
data_mark_r = data_r
for(i in 1:length(data_r[,1])){
for(j in 1:length(data_r[1,])){
if(data_r[i,j] >= 0.8 | data_r[i,j] <= -0.8)
{
data_mark_r[i,j]=round(data_r[i,j], 2)
}
else
{
data_mark_r[i,j]=""
}
}
}
write.csv(data_mark, file=paste(route, "data_mark_p.csv", sep="/"))
write.csv(data_mark_r, file=paste(route, "data_mark_r.csv", sep="/"))
pheatmap(data_r, display_numbers=data_mark, cellwidth=20, cellheight=20, fontsize_number=17, filename=paste(route, "Correlation_result_p.pdf", sep="/"))
pheatmap(data_r, display_numbers=data_mark_r, cellwidth=20, cellheight=20, fontsize_number=7, filename=paste(route, "Correlation_result_r.pdf", sep="/"))
pheatmap(data_r, display_numbers=data_mark, cellwidth=20, cellheight=20, fontsize_number=17, filename=paste(route, "Correlation_result_p.png", sep="/"))
pheatmap(data_r, display_numbers=data_mark_r, cellwidth=20, cellheight=20, fontsize_number=7, filename=paste(route, "Correlation_result_r.png", sep="/"))
}
2 应用
dir.create("metabo_species_0.8r")
select_pheatmap("Correlation_result.txt", "metabo_species_0.8r")


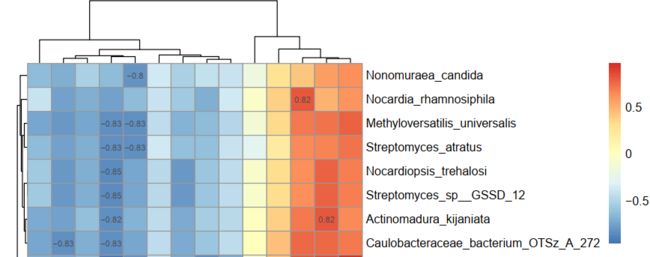
七、cytoscape绘图文件
1 准备文件
# 读取关联分析结果
data = read.table("Result/Correlation_result.txt", sep="\t", header=T)
# 画图文件
data = data[data$raw_p_value <= 0.05,]
r_label = c()
for(i in 1:nrow(data))
{
if(data[i, 3] < 0)
{
r_label = c(r_label, "neg")
}
else
{
r_label = c(r_label, "pos")
}
}
data$r_label = r_label
write.table(data, file="Result/input_pre.txt", sep="\t", row.names=F, quote=F)

data$r_value = abs(data$r_value)
input_network = data[, c(1,2,3,6)]
write.table(input_network, file="Result/input_network.txt", sep="\t", row.names=F, quote=F)

g1 = data.frame(feature=input_network[,1], group=rep("tax", nrow(input_network)))
g2 = data.frame(feature=input_network[,2], group=rep("ko", nrow(input_network)))
input_group = unique(rbind(g1, g2))
write.table(input_group, file="Result/input_group.txt", sep="\t", row.names=F, quote=F)

2 cytoscape绘图
过程见:Cytoscape绘制相关网络图
3 几种常用的layout
layout -> group attributes layout -> group

layout -> group attributes layout -> degree
layout -> attribute circle layout -> degree
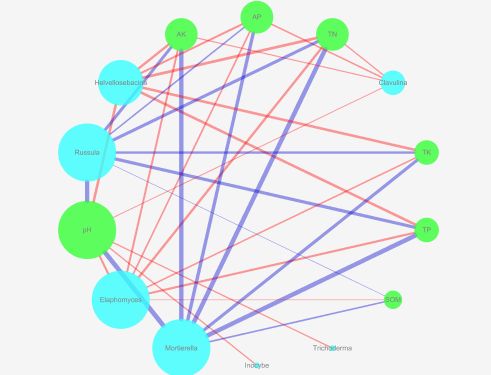
layout -> attribute circle layout -> group

于2020.9.21更新:fdr_p raw_p的对应关系
于2020.9.22更新:输出绘图文件data_p data_r data_mark
于2020.10.14更新:增加了根据R值筛选相关的select_pheatmap