一、多序列比对能干嘛?
下面是多序列比对的主要应用:
1. 推测——Extrapolation
可以推测一条未知的aa序列属于某个已知的蛋白质家族或者拥有相似蛋白质结构域甚至相似的蛋白质3D结构等。
2. 系统发育分析——Phylogenetic Analysis
如果选择合适的序列进行多序列比对,可以分析他们的系统发育关系。比如利用BioWeb(https://bioweb.pasteur.fr/welcome)的Pasteur Phylip(https://evolution.gs.washington.edu/phylip.html)或者Phylogeny.fr等网页上的工具可以实现(http://phylogeny.lirmm.fr/phylo_cgi/index.cgi,这个网站的新版:https://ngphylogeny.fr/ 刚刚朋友M还给我介绍了另一个工具:SMS,如果用PhyML建树的话,可以在SMS上先跑一下,得到建树的推荐参数设定)。

SMS的那篇论文
预测结构,预测功能,构建系统发育树?听上去都是生物研究中需要分析的基础项。那么怎么做?就是将目标序列与数据库中多条同源序列的相似部分摆放在同一栏,同一位置。
有一些工具可以帮助我们进行多序列比对,但是最好再根据结构、进化、功能、序列相似性人工矫正一下比对结果。
二、选择合适的序列
1. 选择序列时要注意的问题
选择要比对的序列很重要,不然结果没有意义。这些序列一般同源、同一家族等。但是同源,同一家族的蛋白质也太多了,我们选择时一般可以注意以下几个方面(比较通用的几点,有特殊实验要求的另外考虑):
(1)一般选择比较蛋白质序列比DNA更好(因为蛋白质序列短而且含有的20种氨基酸信息比DNA有的的4种核苷酸信息更多;如果是非编码区就只能选DNA序列比对);
(2)选择的数据库中的序列最好有一些有详细的注释,这样可以提供很多信息;
(3)多序列比对选用10-15条序列开始比对(如果10条的结果不错,又想再加别的序列进行分析也可以。如果结果不好,需要对现有的序列进行处理,比如删除,剪辑等。比对序列的数量不是越多结果越好,多了反倒增加软件出错概率,除非工作需要);
(4)如果有一条序列与半数以上的其他序列一致性低于30%,比对会有些问题(一般aa序列一致性在30%-70%之间,E-value在10^-40到10^-5,不过这并不是硬性规定);
(5)如果有序列之间一致性太高的,进行多序列比对也没有什么价值(除非有特殊实验目的,具体问题具体分析。需要权衡结果是要能更多地体现相似性还是提供新信息。序列之间高度相似,一定会有很好的比对结果。但是提供的新信息会少);
(6)很多工具善于比对总长度类似的序列,对长短不一的分析结果不好,如果可以,需要提前剪辑;
(7)一般工具对有重复片段的多序列进行比对时存在问题,尤其序列间重复的次数不同时问题更大,需要人工提取这部分,进行分析。
2. 操作示例
以人的钙依赖性肌酶蛋白calcium-dependent kinase proteins—— 序列号为P20472的序列为例。可以在ExPASy、Swiss-Prot、NCBI的blastp页面上直接输入序列号,检索,得到一系列同源序列,再按照上述规则选择合适的多条序列,下载FASTA格式文件。
(1)下面截图是ExPASy-blastp网页的(https://web.expasy.org/blast/),如果选择的序列它们长度相似且不需要额外剪辑,还可以直接勾选发送到ExPASy-Clustal W,进行多序列比对。

输入序列号或者原始序列
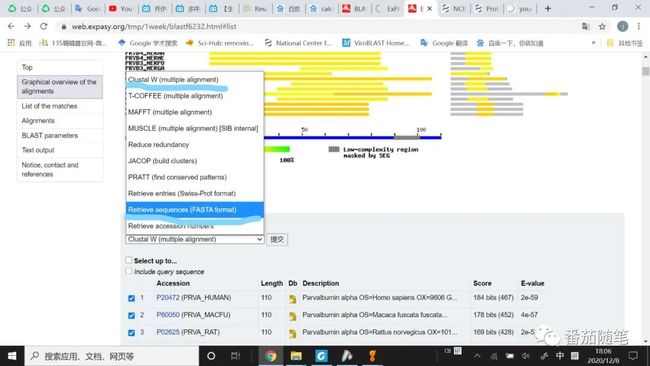
得到多条比对结果,选择并且导出FASTA格式或者直接发送到ExPASy-Clustal W(如下图)

很多网站都有Clustal,MUSCLE的插件,在下面【三、选择合适的多序列比对的方法】中会多介绍,这里是因为ExPASy等网站可以直接将数据发送到多序列比对MSA的页面,就先写了;同样,很多MSA网页又可以直接将比对结果send to系统发育分析的网页。
(2)在UniProt网站使用序列号提取全部序列
如果我们知道自己要比对的多条序列的序列号,可以直接在这里(https://www.uniprot.org/uploadlists/)提取,点击底下的submit即可。

输入序列号

这里除了提取序列,还可以通过序列号直接提取序列的其他信息
三、多序列比对软件/程序
目前常见的有Clustal,、MUSCLE、T-Coffee和MAFFT等。
1. Clustal
由于是第一款多序列比对的软件,所以使用较多,很多网页都有这个的功能插件(比如EBI,EMBnet,PIR,GenomeNet,DDBJ等)。它的更新版本也蛮多,之前是Clustalx,Clustal W系列。现在最新的是Clustal Omega,可最多比对4000条序列/小于4MB的文件。
EMBL-EBI-Clustal Omega
https://www.ebi.ac.uk/Tools/msa/clustalo/
GenomeNet-Clustal W
https://www.genome.jp/tools-bin/clustalw


Clustal Omega算法流程图,整体来讲,Clustal系列采用累进算法(progressive methods)。首先进行序列两两比对,构建距离矩阵→基于两两比对距离矩阵,由关系近的序列逐渐加入关系远的序列构建引导树guide tree→进行多序列比对。由此可见,比对的准确性高度依赖于一开始的两两比对,比较适用于亲缘关系较近的序列。Clustal Omega中改进的新两两比对和建guide tree算法使Omega在W的基础上,速度、准确度和数据处理量上与所提升。
2. MUSCLE(MUltiple Sequence Comparison by Log- Expectation)
https://www.ebi.ac.uk/Tools/msa/muscle/
最多比对500条序列/小于1MB的文件。

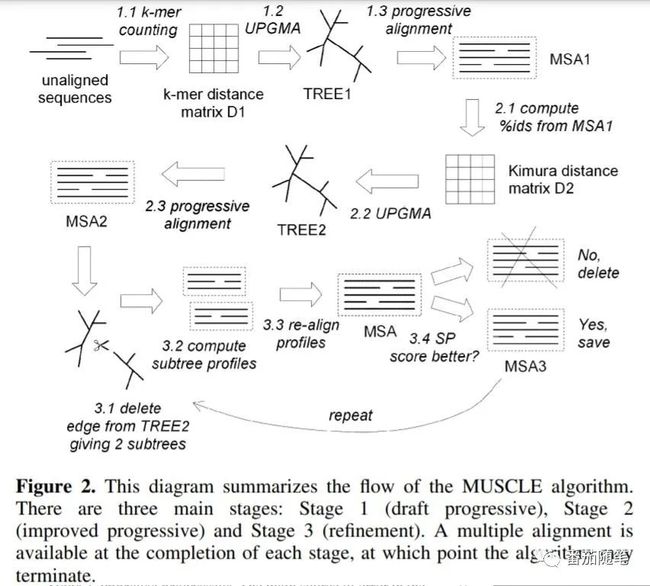
MUSCLE第一篇论文,介绍了算法。整体看也是累进算法,但是在此基础上还有对引导树受限分区进行调整和修正。
3. T-Coffee
最多比对500条序列/小于1MB的文件。
https://www.ebi.ac.uk/Tools/msa/tcoffee/
官网:http://tcoffee.crg.cat/apps/tcoffee/index.html


Coffee系列感觉很不错啊,我是它颜粉。M-Coffee很与时俱进,显示结果会指出其他的软件,比如Clustal,MUSCLE等比对与Coffee结果一致的部分,一致部分比对可信度高,给科研工作者以参考,不用手动去不同平台比对再进行比较了。
Coffee还推出了一个对MSA结果评估的工具TCS:


T-Coffee算法流程图,方形框是操作,圆角框是数据结构。整体上还是累进算法,基于Clustal的算法。
4. MAFFT(Multiple Alignment using Fast Fourier Transform)
https://www.ebi.ac.uk/Tools/msa/mafft/
最多比对500条序列/小于1MB的文件。特定是速度快。

MAFFT中实现了两种不同的算法,即累进方法(FFT‐NS‐2)和迭代优化方法(FFT‐NS‐i)。迭代方法(iterative methods)针对累进比对的不足,在比对过程中不断重新比对各个亚组序列,再把亚组序列重排成包括所有序列在内的整体比对,从而获得最优比对。
若我们在UniProt提取编号为P20472, P80079, P02626, P02619, P43305, P32930, Q91482, P02620, P02622, P02627的蛋白质序列的FASTA格式,上传上述四个网站,结果是差不多的(应该是我找的这几个序列对比太简单了,序列长度都类似)。硬要仔细比较,可能是MUSCLE吧,它的distance矩阵看起来好些。
搜了一下别人的看法,大家都各有惯用的工具。另外有一个上文没有提到的工具“PRANK”在发现多序列保守区域中表现很好,只是速度太慢,不适合较大文件。而且用这些工具进行比对之后,往往还需要手动调整、裁剪等,再进行下一步分析。
用不同的工具进行多序列比对时,还可看看它们能设定的参数。有的明显更加适合你的数据(或者有的数据用什么工具的结果都类似,就像我文中选的这10条序列)。找到自己喜欢的工具和网页,多了解可以设定的参数(一般网站设定参数后面都有小问号解释),更好的进行分析。

MUSCLE的distance matrix
一组简单序列用不同工具的MSA结果:

四、评估多序列比对结果
1. 从结果的显示可以简单看出:
保守程度由高到低为“ *→:→ · ”
* 保守栏,序列一致。
: 保守性突变,那几个氨基酸可能是同种性质的,如分子量,电荷极性等。
· 半保守性突变。
2. Coffee-TCS也可以评估:
从粉到蓝,good→bad

3. 对结果进行进一步分析
我们进行MSA多数是为了找到这些序列的重要片段,重要片段的序列组成保守,不易突变,即使是在亲缘关系较远的序列间也相对保守。
上面的例子中几个蛋白序列的MSA比对结果较好,通过评估只可以看出来N末端比C末端更加保守,推测在N末端更有可能是活性位点。但是范围有些大且究竟是不是,还需引入差异大一些的序列进一步分析。
我们可以在拥有很好比对结果的序列基础上引入兔子(P02586)和老鼠(P19123)的相应钙依赖性肌酶蛋白的序列,再进行一次MSA(P20472, P80079, P02626, P02619, P43305, P32930, Q91482, P02620, P02622, P02627, P02586, P19123)。
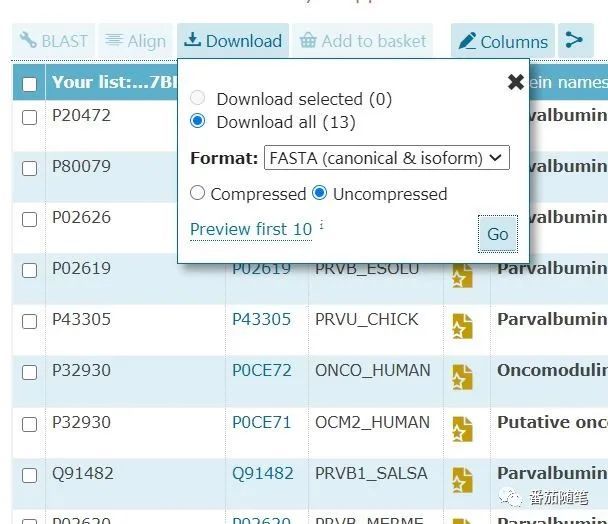
直接提取相关序列并下载

MUSCLE比对加T-Coffee-TCS评估
原来一大块比对的粉色区域被拆开了,可见粉色深的区域更加保守,很可能这些蛋白的活性位点在这个区域。
在Geneious软件上的MUSCLE比对结果图也很直观:绿色identity=100%;军绿色identity>30%;红色identity<30%。引入两条序列后,根据新的MSA结果推测钙依赖性肌酶蛋白的活性位点(钙离子结合位点)在下图的圈里,这个推测也与数据库中的注释信息一致,耶!开心。
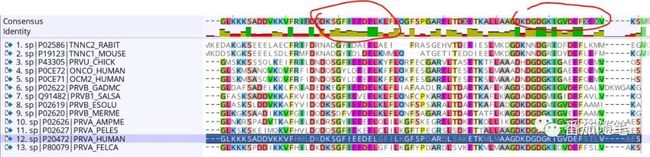
Geneious MUSCLE比对结果

数据库中关于人类钙依赖性肌酶蛋白P20472的功能区域注释,位置和比对结果可以对应
五、在一些unaligned蛋白序列中寻找保守功能域/DNA序列中找蛋白质结合位点(非比对MSA)
有时候我们要比较亲缘关系太远或没有同源性的序列之间相似的部分,或想发现蛋白序列中复杂可变的模体。上面介绍的MSA程序就都不好用了,这时可以试试基于统计学方法的Pratt等工具,用以发现不能比对的序列的保守motif。
Pratt(https://www.ebi.ac.uk/Tools/pfa/pratt/),EMBL上的描述如下:

类似的分析不方便比对的序列的保守motif的工具还有:
Bioprospector (http://ai.stanford.edu/~xsliu/BioProspector/)
Improbizer(https://users.soe.ucsc.edu/~kent/improbizer/improbizer.html)
六、多序列比对总体思路
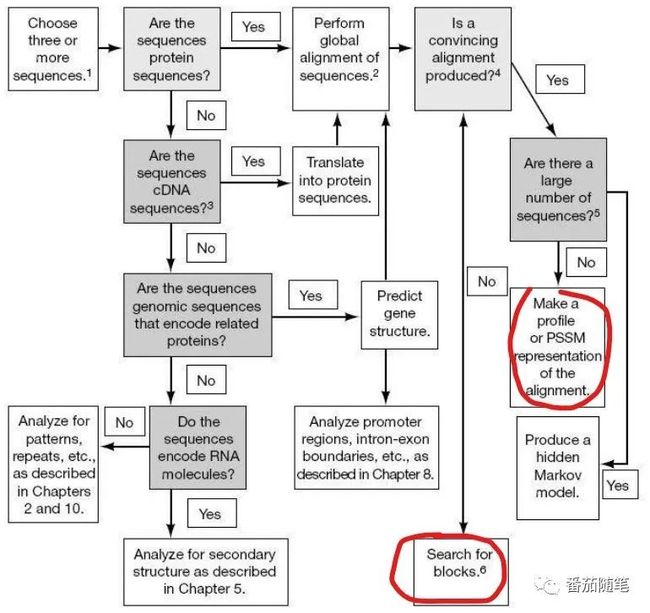
这个图体现了多序列比对总体思路,我在国内外很多讲义上看到,没有注明来源,我也没有找到是哪本书上的。这汇总的很棒,很清晰。今天这个推送里主要介绍的就是这张图中两个红圈方块,上方红圈是比对MSA,下方红圈是非比对MSA。如果小伙伴知道是哪本书的还请留言哈。
这期有点长,给看到这里的小伙伴笔芯♥
如果有错误还请留言哦,共同进步♥
往期相关内容:
【陪你学·生信】序
【陪你学·生信】一、生信能帮我们做什么
【陪你学·生信】二、一些你肯定会用到的生信工具和基本操作
【陪你学·生信】三、核苷酸序列数据库的使用
【陪你学·生信】四、蛋白质相关的数据库
【陪你学·生信】五、当你有一段待分析的DNA序列(基础操作介绍)
【陪你学·生信】六、当你有一段待分析的氨基酸序列(基础操作介绍)
【陪你学·生信】七、在数据库中检索相似的序列
【陪你学·生信】八、序列两两比对