用EnrichmentMap显示富集结果Visualization of enrichment results with EnrichmentMap ●时间~5分钟
7.启动Cytoscape软件。Cytoscape入门教程可以在http://tutorialscytoscape.org中找到。
8.在菜单中,点击Apps→ EnrichmentMap。
9.创建富集图面板将会出现(fig6)。使用g:Profiler(选项A)和GSEA(选项B)创建富集图需要不同的输入文件。

(A)为g:Profiler在步骤6A中生成的结果创建富集图
(i)在Create Enrichment Map面板中,点击folder图标(图6a)。
(ii)找到并选择包含g:Profiler结果的结果数据文件夹,然后单击Open
(iii)在右侧窗格中,g:Profiler输出文件将自动填充到指定的字段中。或者,用户可以单击“+”符号手动指定所需的每个文件。
(iv)在右侧窗格中,如果需要,修改创建的数据集的名称。默认情况下,mentmap将使用g:Profiler富集结果文件的名称(例如,' Supplementary_Table4_gprofiler_results.txt ')。
(v)验证分析类型设置为Generic/gProfiler
(vi)验证富集结果文件是在步骤6A(习)中下载的g:Profiler文件(或者手动指定“Supplementary_Table4_gprofiler_results.txt”)。
(vii)验证指定的GMT是从步骤6A(xii)的g:Profiler网站检索到的文件。使用文件的hsapi .path . name。(或者手动指定“Supplementary_Table5_hsapiens.pathways.NAME.gmt”),其中包含与GO生物过程和反应体路径相对应的基因集。
附加文件说明:
(viii)表达。(可选)上传g:Profiler中分析基因的表达矩阵,或者上传所有基因的表达数据集。如果表达式数据集包含未用于g:Profiler搜索的其他基因,则它们的表达式值仍将出现在富集图的热图中(例如,示例文件,请参见补充表6)。
(ix)级别。(可选)可以指定基因列表或表达数据的级别(例如,参见补充表2)。
(x)类。(可选)这是一个定义表现型的GSEA CLS文件(即,生物条件)中每个样本的表达文件;例如,请参见补充表7。该文件仅用于GSEA中的表型随机化;然而,如果将其提供给EnrichmentMap,则会按表现型在EnrichmentMap heat map viewer中标记表达式文件的列。
(xi)表型。(可选)如果表达数据中有两种不同的表型,更新表型标签,使“阳性”表示与阳性值相关的表型(本例中为间充质),“阴性”表示与阴性值相关的表型(本例中为免疫反应性)。
调优参数
(xii)节点数量。默认情况下,g:Profiler只返回具有统计意义的结果(Q < 0.05),因此,除非需要更严格的过滤,否则在EnrichmentMap输入面板中可以将FDR Q值截止参数设置为1。对于本方案,将FDR Q值设置为0.01。(可选)通过表达式选择筛选基因,以排除基因集定义文件中的任何基因(如,GMT)即在提供的表达式文件中没有找到。如果没有选择通过表达式筛选基因,那么在表达式文件中没有找到的任何基因都将被保留,并显示在表达式热图查看器中,所有相关的表达式值都将显示为灰色。
(xiii)边数。将连接性滑块放在中间。如果网络由于太多的连接(边缘)而过于杂乱,那么将滑块向左移动,使网络更加稀疏。或者,如果网络过于稀疏(即,则将滑块向右移动,以获得连接更紧密的网络
CRITICAL STEP:将滑块向左(或向右)移动将调整基础相似性统计阈值,使生成的网络更稀疏(或更密集)。滑块设置为预定义的默认值,但是用户可以通过选择Create Enrichment Map面板底部的Show advanced选项微调相似性度量。预定义值作为滑块上的滴号出现,包括Jaccard > 0.35、Jaccard > 0.25、组合> 0.375、overlap> 0.5和overlap> 0.25
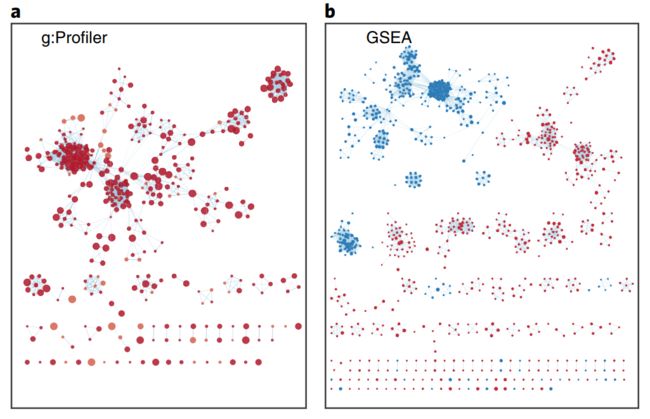
(xiv)点击EnrichmentMap输入面板底部的Build按钮。将出现一个Building EnrichmentMap框,并指示进度状态。一旦成功创建地图,此框将消失。从g:Profiler分析得到的富集图见fig7a.
(B)根据步骤6B产生的GSEA结果绘制富集图:
(i)在Create Enrichment Map面板中,点击文件夹图标,找到步骤6B中创建的GSEA 结果文件夹(fig6)。
(ii)点击GSEA文件夹进行选择。点击打开。
CRITICAL STEP: 如果指定一个包含多个GSEA结果的目录,而不是一个单独的GSEA结果文件夹,mentmap将把每个GSEA结果文件夹视为一个单独的数据集。这使多数据集分析变得容易。如果您只想要一个数据集,但无意中选择了包含多个GSEA结果的目录,而不是选择单个文件夹,那么只需选择不想使用的数据集,然后单击mentmap输入面板顶部的垃圾桶删除它们.
(iii)在右侧窗格中,GSEA输出文件将自动填充到指定的字段中。另外,可以单击“+”符号手动指定所需的每个文件.
(iv)默认情况下,EnrichmentMap将在最后一个点(.)之前使用GSEA文件夹名称的第一部分来创建数据集名称。例如,如果目录名为Mesen_vs_Immuno。GseaPreranked。名称将填充为Mesen_vs_Immuno.GseaPreranked.
(v)验证分析类型设置为GSEA。
(vi)富集 Pos.(阳性)验证文件名设置为' [your_path_to_gsea_dir]/ Mesen_vs_Immuno.GseaPreranked.12345/gsea_report_for_na_pos_12345]。,其中“12345”是GSEA生成的唯一数字。或者导航到“Supplementary_Table8_gsea_report_for_na_pos”。xls”文件
(vii)富集 Neg。(阴性)验证文件名是否设置为' [your_path_to_gsea_dir]/ gsea_report_for_na_neg_12345。,其中“12345”是GSEA生成的唯一数字。或者导航到文件“supplementary_table9_gsea_report_for_na_negg .xls”。
(viii)GMT。验证文件名是否设置为“supplementary_table3_human_gobp_allpathways_no_go_iea_july_01_2017_symbol .gmt”。或者,导航到文件“Supplementary_Table3_Human_GOBP_AllPathways_no_GO_iea_July_01_ 2017_symbol.gmt”
(ix)排序。验证文件名是否设置为'ranked_gene_list_na_pos_versus_na_neg_12345。其中“12345”是GSEA生成的唯一数字。或者,导航到“Supplementary_Table2_MesenvsImmuno_RNASeq_ranks。rnk”文件。
附加文件说明:
(x)表达。(可选)上传GSEA中分析基因的表达矩阵。为
示例文件见“Supplementary_Table6_TCGA_OV_RNAseq_expression.txt”。
(xi)类。(可选)这是一个定义表现型的GSEA CLS文件(即、生物表达式文件中每个示例的条件。例如,请参见“Supplementary_Table7_TCGA_OV_RNAseq_classes.cls”。“这个文件只适用于表现型在GSEA随机化;然而,如果将其提供给EnrichmentMap,则会按表现型标记富集映映射图表达式文件的列。
(vii)表型。(可选)文本框中的“na_pos”替换为“Mesenchymal”和“na_neg”
替换为“Immunoreactive”。“Mesenchymal”将与红色结节相关联,因为它对应于阳性表型,而“Immunoreactive”表型将被标记为蓝色。
CRITICAL STEP :如果在指定显型之前加载CLS文件,EnrichmentMap将自动从类文件中猜测显型。如果您的类文件指定了两个以上的显型,EnrichmentMap将选择文件中定义的前两个显型。要在EnrichmentMap heat map中注释表现型,指定的表现型标签需要与GSEA CLS文件完全匹配。
参数调优
(xiii)节点数。将FDR Q值截止值设置为0.01。(可选)通过表达式选择筛选基因,以排除基因集定义文件中的任何基因(如:GMT)即在提供的表达式文件中没有找到。如果没有选择通过表达式筛选基因,则在表达式文件中没有找到的任何基因将保留,并将在表达式查看器中显示其所有相关的表达式值.
将连接性滑块放在中间。要创建边缘更少的网络(稀疏网络),请将滑块向左移动。或者,要创建边缘更多的网络(密度更大的网络),将滑块移动到右边。
(xiv)边数:将滑块向左(或向右)移动的关键步骤将调整基础相似性统计阈值,使生成的网络更稀疏(或更密集)。滑块设置为预定义的默认值,但是用户可以通过选择Create Enrichment Map面板底部的Show advanced选项微调相似性度量。预定义值作为滑块上的滴号出现,包括Jaccard > 0.35、Jaccard > 0.25、overlap> 0.375、overlap> 0.5和overlap> 0.25
(xv)点击EnrichmentMap输入面板底部的Build按钮。一个Building将出现EnrichmentMap框,并指示进度状态。一旦成功创建了映射,这个框会消失.得到来自GSEA的分析的富集图见图7b.
丰富图的导航和解释Navigation and interpretation of the enrichment map 时间 约 4 小时
CRITICAL :必须解释富集映射,以发现关于数据集的新信息,并且必须手动细化,以创建高质量的公开图。
10 .要查看富集图,请在Cytoscape窗口左侧的控制面板中选择感兴趣的网络(图8),可以使用控制面板最左侧的选项卡选择网络面板。所选网络将出现在主窗口;使用细胞角控件导航到它(缩放和平移)(图8 (i)),并通过阅读基因集标签探索路径。具有许多共同基因的通路通常代表类似的生物学过程,并被组合成网络中的子网络或主题。点击一个节点,在网络视图下的表格中显示相应的基因(图8 (ii))。
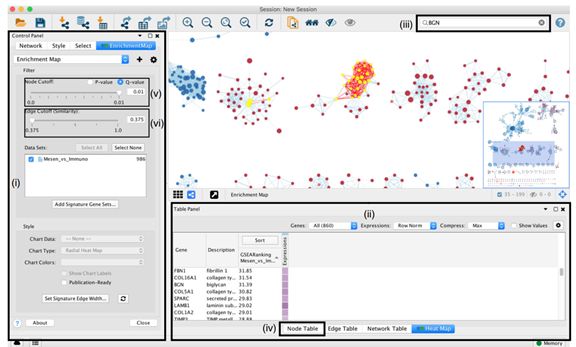
11.要找到感兴趣的基因或途径,请在位于右上角的搜索栏中键入其名称(图8 (iii))。所有包含该基因的通路都将被突出显示。例如,在g:Profiler和GSEA分析中,TP53和BGN分别是最主要的基因。
12.要找到最大富集的路径,请查看位于network视图下方的表面板。选择Node Table选项卡(图8 (iv)),单击列标题,选择并排序名为' EM#_fdr_qvalue '(用于g:Profiler)或' EM#_NES '(用于GSEA)的列。要突出显示网络中这些路径的子集,请选择与感兴趣的路径对应的行,右键单击表中任何选定的行,并从选定的行中选择select nodes。
13.根据分析的重点,可以对生成的富集图采取不同的操作。按照选项A查看表面板热图;选项B组织和阐明网络;选项C定义主要的生物主题;选项D创建一个简化的网络视图;选项E手动排列网络节点;选项F创建一个突出显示特定主题子集的子网络。跳过第14步保存图像并生成图例。
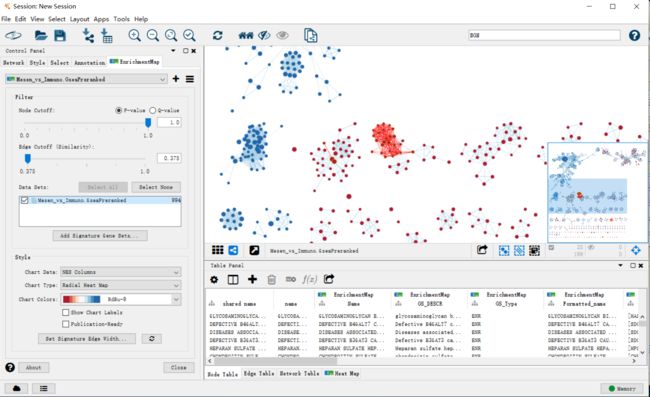
(A)探索表面板热图 ● 时间45分钟
(i)当基因表达矩阵作为富集图谱的输入时,我们可以研究富集通路中包含的基因的表达模式。点击单个节点或一组节点生成一个基因表达热图,将出现在表的热图选项卡面板(fig9)。如果分析是基于GSEA结果和排名文件提供,“前沿”的基因会以黄色突出显示单个节点的选择。有几种热图可视化选项。
CRITICAL:如果没有提供给EnrichmentMap表达文件作为输入,它会自动创建一个虚拟表达文件在富集文件中发现的任何基因表达将被赋予一个占位符表达式值为0.25,和任何在通路中找到的基因,但在富集的结果文件没有发现的将被分配一个占位符表达式值“NA”(不适用)。因此,单击富集图中的任何节点将显示用于分析的基因,以及不属于查询集的路径中的基因.

(ii)调整排序选项。排序选项包括层次聚类、等级或无排序。要更改排序选项,请单击热映射表左上角可见的Sort按钮(fig9 (i))。默认情况下,如果提供了秩文件,则按秩对热图进行排序.如果没有rank文件,则不应用排序。可以通过热图面板右上角的设置菜单(fig9 (vi))上传其他等级文件进行比较。通过单击sort按钮并按名称选择rank文件,可以选择要排序的rank文件。单击当前排序列旁边的箭头以反转排序顺序。单击任何列名对所选列进行排序。
(iii)定义您希望在热图中可视化的基因(fig9 (ii))。可以查看所选节点中包含的所有基因(节点的联合)的数据,也可以只查看所选节点的公共基因(节点的交集)的数据。默认情况下,显示所有的基因。
(iv)根据数据类型更改表达值的可视化(fig9 (iii))。可以将数据视为已加载的数据(值)、行规范化的数据(在这种情况下,行平均值从每个值中减去,然后除以行标准差(行范数),或者是对数转换的数据(Log -transformed, Log)
(v)压缩热图列(fig9 (iv))。默认情况下,对于小于50个示例的表达式集,所有表达式值都作为热图中的单独列可见。通过选择压缩下列出的聚合方法之一(中值、最大值或最小值),可以将数据压缩到单个列中。如果已经上载了CLS文件,则可以使用Class选项对每个定义的示例组使用一列压缩表达式集。如果表达式矩阵包含≥50个样本,EnrichmentMap默认情况下会自动将这些值压缩到它们的中值.
(vi)检查数值(图9 (v)),除热图颜色比例尺外,还显示数值表达
(vii)使用设置面板对热图进行额外的微调,点击cog图标即可访问(图9 (vi))。这包括添加新的秩文件、将热图数据导出为以制表符分隔的文本文件或PDF图像、更改分层集群的距离度量或打开节点表的热图自动对焦等功能。得到的热图如图9所示。在这个图中,使用GSEA rank文件对基因进行排序,用黄色突出显示前沿基因。显示所选节点中包含的所有基因,表达值行规范化,不进行压缩,不显示单个表达数值。列标题根据样本表现型着色。红色为第一表型(间质),蓝色为第二表型(免疫反应性)。
(viii)可以将热图导出为文本文件进行进一步分析:点击热图的设置图标(图9 (vi)),选择Export as TXT。
(ix)如果只选择一个单独的节点,对话框将只提供导出前沿用于GSEA分析。如果选中,只输出突出显示的基因;否则,整组基因就会被保存下来。
(x)指定文件名和位置,然后单击Save。
(B)网络的组织和解释:时间30分钟
(i)如果网络节点太多,转到控制面板的EnrichmentMap选项卡,使用节点截止q值阈值滑块。调整到接近于0的数值将删除不太重要的节点(图8 (v))。
(ii)如果网络连接过于紧密,请转到控制面板的mentmap选项卡,并增加边缘截止(相似性)阈值;这将删除关联较少的节点之间的连接(fig8 (vi))。
(iii)调整好分割线后,再次应用网络布局(参见Cytoscape中的布局菜单Layout)。默认的布局算法是未加权的级联力定向布局(Prefuse Force Directed layout)。我们还建议使用基因集相似性系数对力导向布局进行加权。其他布局算法是可用的,我们鼓励使用它们进行试验。
CRITICAL STEP:Cytoscape中有许多不同的布局算法,可以用EnrichmentMap。
我们建议使用边缘加权布局,它考虑路径之间的重叠评分。大多数布局(除了yFiles)提供仅组织所选节点的能力。尝试不同的布局,看看哪种布局最适合您的数据。如果您不喜欢最终的布局,在macOS上按command-z或在Windows上按Ctrl-z,或者单击Edit→Undo恢复到以前的视图。
(iv)若要还原节点或边缘,请将阈值滑块调整到其原始位置。
(v)分离两种不同的表型。将两种不同的表型(即,将所有红结点放在一边,所有蓝结点放在另一边)。为此,转到控制面板中的Select选项卡(fig8 (i))。
(vi)点击“+”符号,选择“列过滤器”。
(vii)点击“选择”列,然后选择“EM1_NES (Mesem_vs_Immuno)”。
(viii)点击between旁边的框,将值更改为0。单击面板底部的Apply。
(ix)现在应选择所有红色节点。单击并按住所选的任何节点,并将所选内容拖到左边,直到它不与任何蓝色节点重叠为止。
(x)从Cytoscape菜单中选择Layout,应用Prefuse Force Directed Layout 定向布局→只选择节点→(none)。
(xi)返回控制面板选择选项卡,调整滑块以选择所有负值。
(xii)现在应该选择所有蓝色节点。单击并按住所选的任何节点,并将所选内容拖到右边,直到它不与任何红色节点重叠为止。
(xiii)从Cytoscape菜单栏中选择Layout,应用Prefuse Force Directed Layout定向布局→只选择节点→(无)
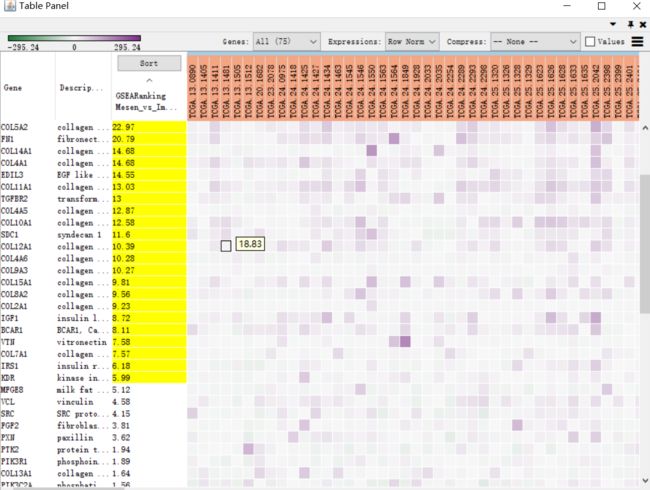
(C)确定主要的生物学主题:timing2.5小时
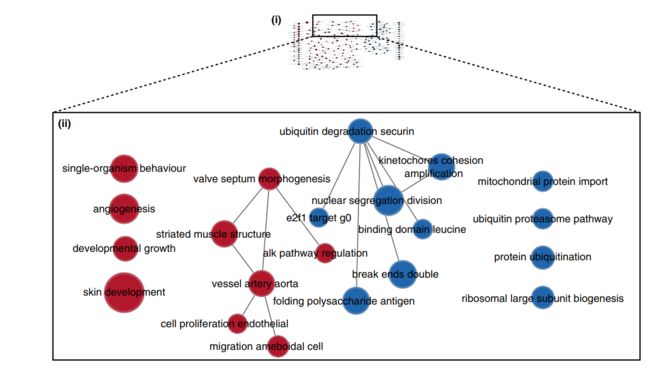
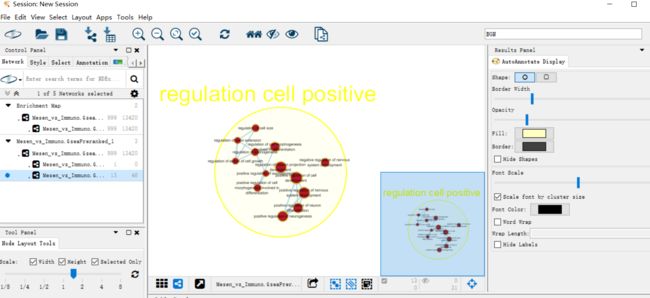
CRITICAL富集图谱通常包括代表主要生物主题的类似途径的簇。可以使用AutoAnnotate Cytoscape应用程序自动定义和总结集群。AutoAnnotate首先使用clusterMaker2应用程序对网络进行集群,然后通过WordCloud应用程序根据通路名称中的单词频率总结每个集群。
(i)在Cytoscape菜单栏中,通过选择Apps→AutoAnnotate→New Annotation Set启动AutoAnnotate,将出现AutoAnnotate: Create Annotation Set面板。
(ii)在Quick Start选项卡中,单击Create Annotations。
(iii)网络中的每个集群周围都将绘制一个圆圈注释,并将与一组单词(默认为三个)相关联,这些单词对应于集群中最常见的节点标签。这些词是自动选择的,通常必须手动重命名(步骤13E(iii))。在集群中移动单个节点将自动调整周围的圆的大小,而移动整个集群将在新的集群位置中重新绘制周围的圆。
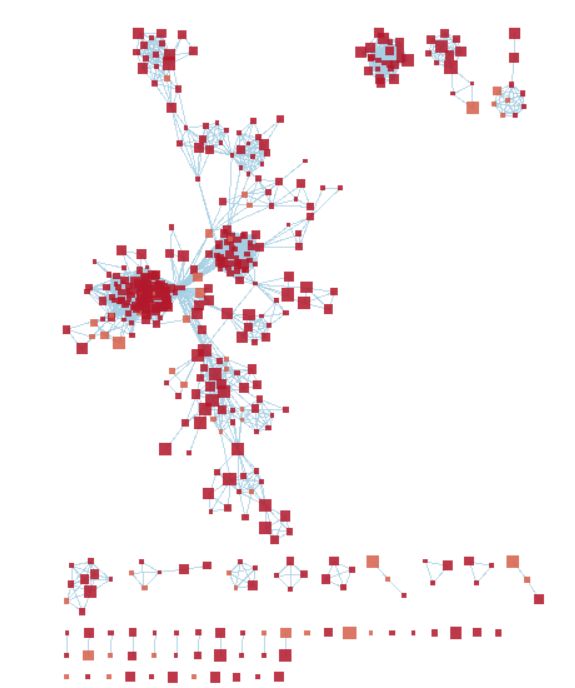
(iv)手工安排集群整理图形。移动节点以减少节点和标签重叠。fig10显示了这个过程的结果。
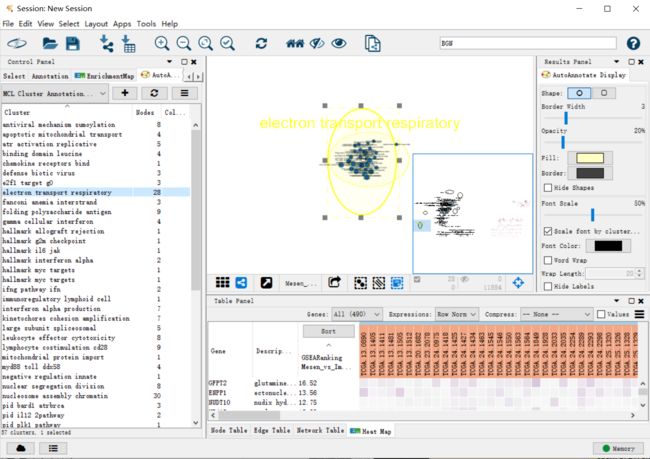
(D)创建一个简化的网络视图:Timing15分钟
CRITICAL为每个集群创建一个具有摘要名称的单组节点,并提供了包含许多节点的富集通路有用于的富集结果主题的概述(fig11)。
(i)在控制面板中,选择AutoAnnotate选项卡。
(ii)点击右上角的菜单图标。
(iii)选择全部折叠。
(iv)缩放倒塌的网络,以便更好地观看。在Cytoscape菜单栏中,选择View→Show Tool Panel。
(v)进入控制面板底部的节点布局工具面板。
(vi)定位缩放滑块,并在未选中的节点上使用(只选中未选中)。
(vii)向左移动滑块,收紧节点间距。完成后关闭节点布局工具面板。

(E)手动布置网络节点,更新主题标签:Timing45分钟
CRITICAL此部分用于最清晰的网络视图和发布质量图。例如,把类似的主题放在一起是很有用的,比如信号或代谢途径,即使它们在地图上没有连接。应该优化空间的使用,以避免出现大量空白。这是一个耗时的步骤,但是花费的精力越多,得到的图的质量就会越高(fig10)。
(i)如果图形的焦点只集中在网络的一个子集上,那么只处理这个子集会更容易。为此,选择感兴趣的节点,然后在Cytoscape菜单栏中选择File→New→Network→From selected nodes→all edges,或者在Cytoscape tools菜单栏中使用相应的图标(New Network From selection→all edges)。
(ii)当该图的目的是显示一个大型网络并仅突出显示主要主题时,单击位于控制面板的Enrichmentmap底部的Publication ready,删除节点标签。要还原到原始网络,再次单击publish ready按钮。
(iii)重命名AutoAnnotate生成的主题名称,以更好地解释路径组。自动生成的wordcloud主题名称对于快速浏览内容丰富的地图很有用,但是经常需要为出版质量的数据重命名。命名应该仔细考虑每个主题中的所有通路和基因。主题可以在AutoAnnotate中重命名,右键单击“集群”列中的AutoAnnotate面板中的名称,然后选择Rename…或者,可以在外部绘图软件中更改标签(按照步骤14-16导出一个文件以在外部绘图软件中使用)。注意,重新集群网络将重置主题名称。
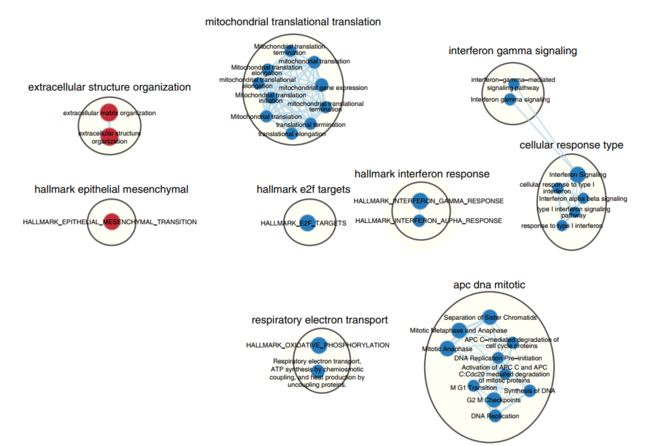
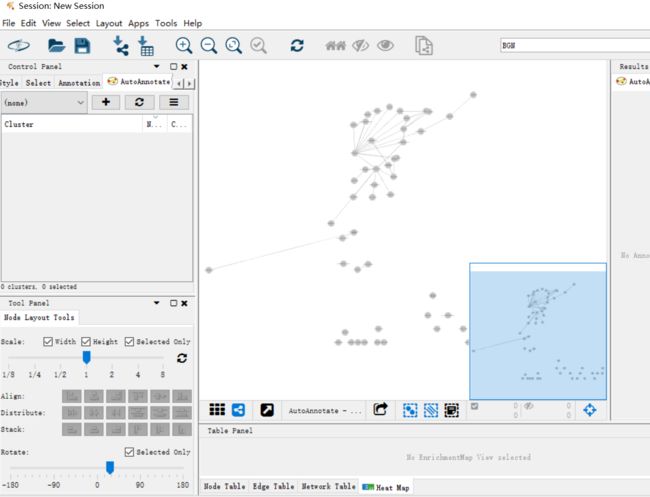
(F)创建突出显示特定主题子集的子网:Timing10分钟
例如,我们将选择最顶端的间充质和免疫反应通路,并为详细的可视化创建子网络。
(i)点击控制面板中的Select选项卡(fig8 (i))。
(ii)点击“+”符号,选择“列筛选器”。
(iii)点击select列,选择EM1_NES (Mesem_vs_Immuno)。
(iv)点击between旁边的方框,将负值替换为“2.5”。不要在包容性旁边更改正值。单击Enter。
(v)点击“+”符号,选择列过滤器。
(vi)点击select列,选择EM1_NES (Mesem_vs_Immuno)。
(vii)点击包容性旁边的框,将值更改为“-2.5”。不要在中间改变负数。单击Enter。在刚刚添加的两个列过滤器上面,将下拉选项从Match all(和)更改为Match any(或)。
(ix)点击应用。在Apply按钮下,它应该显示选中的节点和边缘的数量。在本例中,应该选择32个节点。
(x)从Cytoscape菜单中,选择Select File→New→Network→From selected nodes, all edge。
(xi)一个新的,更小的网络应该出现。手动移动节点来优化的布局。
(xii)按照步骤13C对网络进行注释(fig12)
导出图形,创建图例和保存工作 Exporting figures,creating legends and saving work
14.导出图像。在Cytoscape菜单栏中,选择File→Export as Image…设置导出文件格式为PDF (*. PDF)。
CRITICAL STEP 推荐使用基于关键步骤向量的PDF和SVG格式来处理具有发布质量的数据,因为它们可以在不降低质量的情况下进行缩放。可以使用Adobe Illustrator或Inkscape等软件包编辑这两种文件类型。PNG文件格式推荐用于高质量的在线图像,而JPG格式不推荐,因为它可能会导致有损压缩的视觉工件。
15.点击Browse…指定文件名和位置。
16.单击Save关闭浏览器窗口,然后单击OK.
17.识别网络创建参数。在前面的步骤中,我们将网络导出为图像,但是需要用于创建映射的参数来解释和复制图像。在控制面板中,找到EnrichmentMap输入面板,并单击右上角的cog (Settings)图标。
18.单击“显示创建参数”。在显示的面板中,您将发现要添加到图的文本图例中的FDR Q值、相似性度量和阈值参数。在本例中:‘以参数q < 0.01创建富集图谱,Jaccard重叠组合系数> 0.375,组合常数= 0.5’。
CRITICAL STEP EnrichmentMap创建参数面板只显示用于网络创建的参数。如果使用过滤器或EnrichmentMap滑块修改网络,则必须相应地更新阈值。
19.展示并创造一个图例。在控制面板中,找到EnrichmentMap输入面板并单击右上角的cog图标。单击“显示图例”。示例显示了一个简化的图例;但是,可以在富集映射中使用许多不同的节点和边缘属性,比如大小、形状和颜色,来表示数据的不同方面。重要的是要在与图形相关的文本中描述它们,或者将图形本身描述为图例。图13显示了可用于富集映射图的基本图例组件(在http://baderlab.org/Software/EnrichmentMap#Legends中可以看到SVG和PDF图像)。
20.将所有工作保存为Cytoscape会话。在Cytoscape菜单中,选择File→Save As…导航到您希望保存会话的目录,并指定所需的文件名。
故障排除 Troubleshooting
Troubleshooting advice can be found in Table 1. 故障排除建议见表1。详细见参考文献。
时间Timing
此方案所需的时间主要取决于手工管理的级别和在富集图的可视化组织上花费的时间。较小的网络通常更容易组织。在制定最终的网络之前,有必要重新检查富集分析,以确保其正常工作,充分探索网络,并在最终的发布质量数据中选择要强调的部分。上面的示例分析是在一台拥有8gb RAM和Java 8的Windows 7机器上执行的。增加RAM和处理器速度将减少某些步骤的分析时间,特别是GSEA分析(步骤6B)。
步骤1-5,软件安装:5分钟
步骤6A,使用g:Profiler: 3 min对基因列表进行通路富集分析
步骤6B,利用GSEA对a序列基因表进行通路富集分析:~ 20min
步骤7-9,富集图显示富集结果:~ 5min
步骤10-13,富集图导航与解释:~4 h
步骤13A,浏览表格面板热图:45分钟
步骤13B,组织和澄清网络:30分钟
步骤13C,确定主要的生物主题:2.5小时
步骤13D,创建一个简化的网络视图:15分钟
步骤13E,手工布置网络节点,更新主题标签:45分钟
步骤13F,创建一个突出显示特定主题子集的子网络:10分钟
步骤14-20,导出图形,创建图例和保存工作:15分钟
预期的结果 Anticipated results
利用g:Profiler对基因列表进行通路富集分析 Pathway enrichment analysis of a gene list using g:Profiler
分析中产生了两个主要的示例文件:
●Supplementary_Table4_gprofiler_results.txt。该文件从步骤6A(xi)中的g:Profiler网站下载,包含g:Profiler分析的一组丰富路径,作为一个包含六列的表。第一列是“GO.ID ',包含路径标识符。第二列“description”包含通路描述。第三列,p.Val’,包含该通路的富集P值。第四列,“FDR”,包含了该通路的FDR调节富集P值。g:Profiler只提供fdr调整后的P值,因此第三和第四列中的值是相等的。第5列“表现型”包含一个“1”,表示分析属于阳性表现型。来自g:Profiler的结果总是将表现型指定为1,但是用户可以手动更改列,以便将两个不同的结果集合并在一起。第六列“Genes”包含一个逗号分隔的基因列表,属于该通路。
●Supplementary_Table5_hsapiens.pathways.NAME.gmt。该文件从步骤6A(xii)中的g:Profiler网站下载,包含用于g:Profiler分析的一组路径。文件的每一行表示一个通路,其中第一列是通路名称或标识符,第二列是通路描述,任何后续列都是通路的一部分基因。
利用GSEA对序列基因列表进行通路富集分析 Pathway enrichment analysis of a ranked gene list using GSEA
分析中产生了两个主要的示例文件:
●Supplementary_Table8_gsea_report_for_na_pos.xls。这是主要的GSEA结果表,列出了与na_pos表型相关的通路(在步骤6B(xv)中进行了解释)。
●Supplementary_Table9_gsea_report_for_na_neg.xls。这是主要的GSEA结果表,列出了与na_neg表型相关的通路(在步骤6B(xv)中解释)。
这两个文件都包含11列的表,描述了与每种表型相关的通路的富集程度。有关表中所有值的详细描述,请参见https://software.broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html(在“Detailed Enrichment Results”标题下)。GSEA将创建一个完整的结果文件目录。除非另有说明,否则GSEA结果文件夹将放在gsea_home/(当前日期)下的主目录中。该文件夹将使用步骤6B(xi)中指定的名称命名(例如,根据该协议,' Mesen_vs_Immuno '),并在末尾附加一个13位的随机数。要查看分析的可视化摘要,可以使用web浏览器打开“索引”。在结果目录中找到的html '文件。步骤6B(xiv,xv,xvi),以及图4,5,描述了这些结果的解释。
将通路富集结果可视化为富集图 Visualization of pathway enrichment results as an enrichment map
利用g:Profiler和GSEA分析得到的示例富集图如fig7a、b所示(可公开的GSEA结果见fig10)。通路信息本身是冗余的,因此富集分析常常强调同一通路的多个版本。将冗余路径折叠成一个单一的生物学主题可以简化解释(fig11)。富集图是表示富集通路之间重叠的网络,这些重叠的通路用圆圈(节点)表示,并根据连接通路共享的基因数量用大小相同的线(边)连接。多个结果集可以同时显示在一个富集图中,在这种情况下,不同的颜色用于不同分析产生的颜色路径。如果加载了基因表达数据,点击一个pathway节点,将会显示该通路中所有基因的基因表达热图。
富集图的导航和解释 Navigation and interpretation of the enrichment map
富集图有助于识别组学数据的有趣路径和主题特征。通过确定预期的主题,如癌症基因组学分析中的生长相关通路,可以验证该分析。预期的主题是根据所研究生物系统的专家知识确定的。其他途径被评估为潜在的发现。检测到的通路中的基因应使用原始组学数据和通路图进行解释,即基因相互作用网络或转录因子调控网络。最后,可以发布富集图来支持科学结论,或用于为后续实验生成假设。通路富集分析结果可以根据所使用的参数(如最小和最大通路大小或选择的通路数据库),因此,应该通过改变这些参数来测试结论的稳健性。
报告总结 Reporting Summary
有关实验设计的进一步信息可以在与本文链接的《自然研究报告摘要》中找到。
Data availability 数据可用性
协议使用公共可用的软件包(GSEA v.3.0或更高版本,g:Profiler, EnrichmentMap v.3.0或更高版本,Cytoscape v.3.6.0或更高版本)和应用公共可用的R包(edgeR、Roast、Limma、Camera)的自定义R脚本。自定义脚本可以在补充协议和我们的GitHub网站(https://github.com/baderlab/cytoscape_workflows/tree/master/mentmappipeline和https://baderlab.github.io/Cytoscape_workflows/EnrichmentMapPipeline/index.html)中获得。
参考文献 References
详细见参考文献
致谢 ACKNOWLEDGMENTS
作者感谢J. Mesirov对手稿的评论。该项目由安大略省癌症研究所的J.R.研究员奖和加拿大自然科学与工程研究理事会(NSERC)发现基金资助(RGPIN-2016-06485)。这项工作是由美国国立卫生研究院资助的P41 GM103504, R01 GM070743, U41 HG006623和R01 CA121941到G.D.B.
作者的贡献 Author contributions
美国,教授,这篇手稿是由美国医学博士和博士写的。除了g:Profiler (J.R.)之外,R.I.创建了循序渐进的协议、图表、R脚本和R笔记本。C.T.-L。开发了EnrichmentMap 3.0和AutoAnnotate Cytoscape应用程序。lM.M.,成员j.w.李从先vv测试了这个方案。所有的作者都阅读并批准了最终的手稿。
相互竞争的利益
作者声明没有相互竞争的利益
额外的信息
本文的补充信息可通过https://doi.org/10.1038/s41596-018-0103-9获得。
重印和权限信息请访问www.nature.com/reprinting。
信件和索取资料的要求应寄给G.D.B.
出版者注:施普林格性质对出版地图和机构附属机构的管辖权主张保持中立
相关链接
使用此方案的关键引用
Pinto等人。Nature 466, 368-372 (2010): https://doi.org/10.1038/nature09146
Pajtler, K. W.等人。肿瘤细胞27,P728-P743 (2015): https://doi.org/10.1016/j.ccell.2015.04.002
Cavalli,F. M. G.等。《癌细胞31》,P737-P754 (2017): https://doi.org/10.1016/j.ccell.2017.05.005
学习复现笔记:
step6A
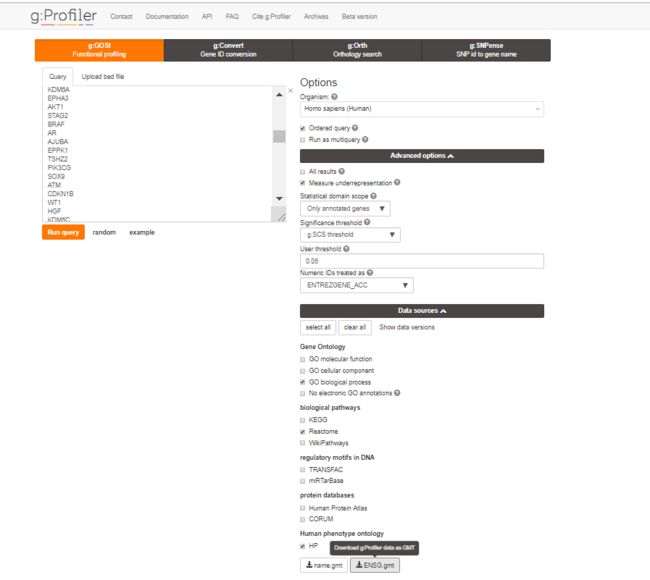
结果复现,step6A,不知道步骤对不对。
step6B
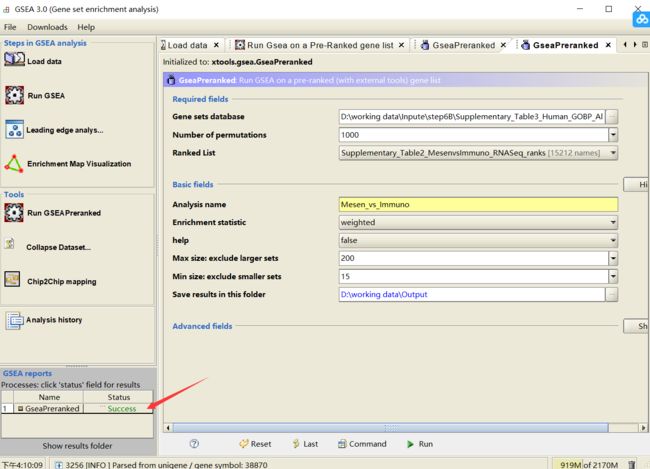


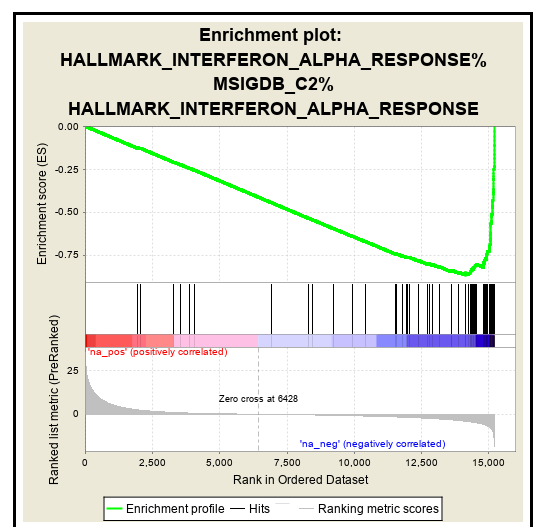
step7A
step7B


step10
step13A
step13B

step13C
step13D

step13E
结果不断学习中。。。。。。
GMT,文件:
用于标准GMT格式的通路富集分析的通路基因集数据库,可从http://baderlab.org/GeneSets下载。该文件在2017年7月1日下载包含从8个数据来源的通路:GO、Reactome、Panther、NetPath、NCI79、MSigDB curated gene sets (C2 collection,不包括Reactome和KEGG)、MSigDB Hallmark (H collection)和HumanCyc。可以从http://baderlab.org/GeneSets获得每月更新一次的基因集。GMT文件是一个文本文件,其中每一行代表一个单一通路的基因集。每一行都包含一个通路ID、一个名称和以制表符分隔的格式列出的相关基因。