R语言包ggplot2绘制多组箱线图
本文记录于2021年6月28日,2021年8月20日更新
最近在做cibersort浸润分析,在对结果做可视化的时候遇到一点问题(问题解答蓝字部分)
需要的代码和数据参考这个资源:https://download.csdn.net/download/weixin_52730798/21402082?spm=1001.2014.3001.5501
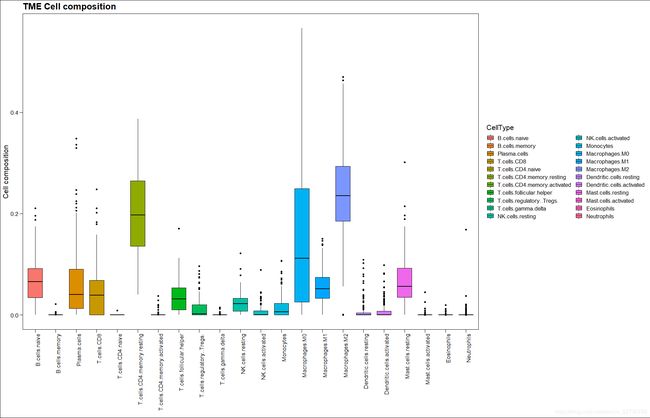
根据默认的绘图只能进行一组数据的绘制,而我想要的结果是不同组数据同时展示
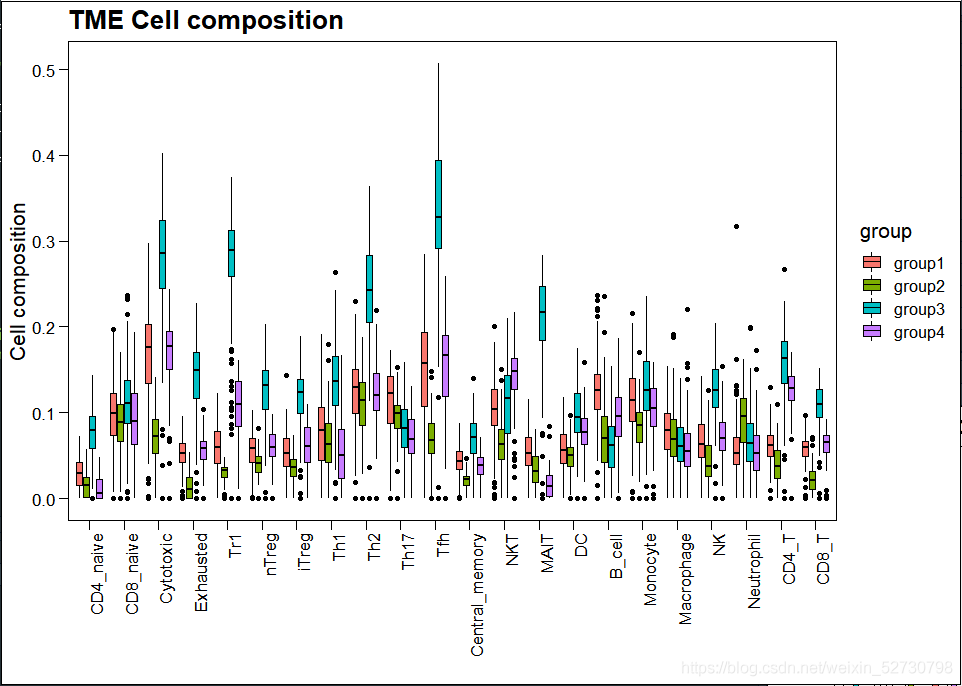
就是这张图这种,然后在网上查了一下这个问题,虽然R语言相关的可视化问题挺多,但是没有发现这个问题怎么解决。
不过也有一些帖子给了启发,就是导入一个额外的样本表型信息,然后再在代码的fill属性中设置选择样本信息,这对和我一样的小白们讲是比较麻烦的一种解决方法。
后来结合这个思路,我找到了一个解决办法:
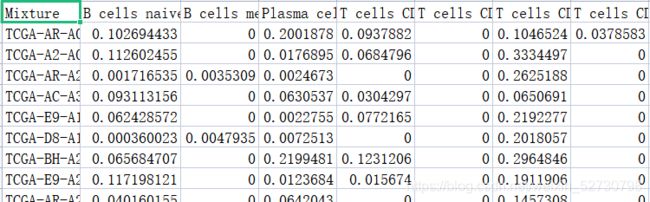
这个是原始数据
这个是自己手动添加了分组信息之后的数据
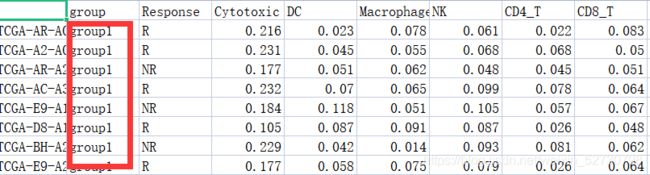
最简单的解决方法就是在数据中对样本直接添加分组信息,根据自己做分析时的分组,直接添加在cibersort的结果中。
代码也只需要非常简单的修改
plot.info_all <- plot.info %>%
as.data.frame() %>%
rownames_to_column("sample") %>%
pivot_longer(cols = 3:8,
names_to = "CellType",
values_to = "Composition")
plot.info_all <- plot.info_all[,c(19,2,20)]
ggboxplot(
plot.info_all,
x = "CellType",
y = "Composition",
color = "black",
fill = "group",#只需要修改这里,讲fill = "CellType"改为fill = "group"
xlab = "",
ylab = "Cell composition",
main = "TME Cell composition"
) +
theme_base() +
theme(axis.text.x = element_text(
angle = 90,
hjust = 1,
vjust = 1
))Q&A
1,针对代码“plot.info_all[,c(19,2,20)]”中的“19,2,20”进行一个解释
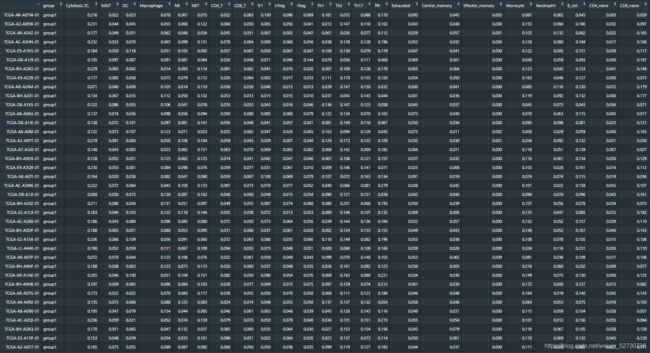
上面这张图是免疫浸润的结果,这个结果是每个样本的每种免疫细胞的评分,对于每个样本,这些评分的和是1,我们进行免疫浸润的目的就是得到这个占比,以此来进行针对单样本层面的免疫分析。
绘图则是对结果进行可视化的展示,在刚才那行代码中,就是对需要展示的结果进行选择,同时需要结合前面一部分代码:
plot.info_all <- plot.info %>%
as.data.frame() %>%
rownames_to_column("sample") %>%
pivot_longer(cols = 3:10,
names_to = "CellType",
values_to = "Composition")注意这部分代码里的“cols = 3:10”,意思是选择刚才那个免疫浸润结果里的第3到10列进行读取
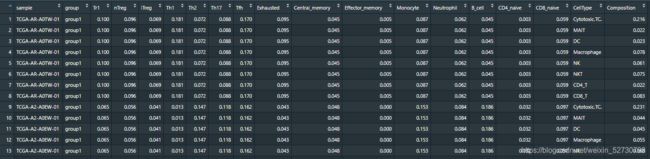

从结果中可以看出,进行列选择之前是24列,选择之后是19列,“19,2,20”中19指的是CellType,就是盒图结果的X轴名称,2指的是分组依据,也就是前面提到的手动添加的分组信息,20指的是Compostion列,也就是将选择的细胞进行计算之后得到的分值
这里多说一句,plot.info_all的数据是以列的方式进行保存的,在绘图时进行读取并绘图的依据便是前面选择的三个对象
如果绘图时想要根据自己的需求进行选择对象,就可以通过这两处修改进行选择了
2,对两组或任意组直接的差异关系进行对比,并显示
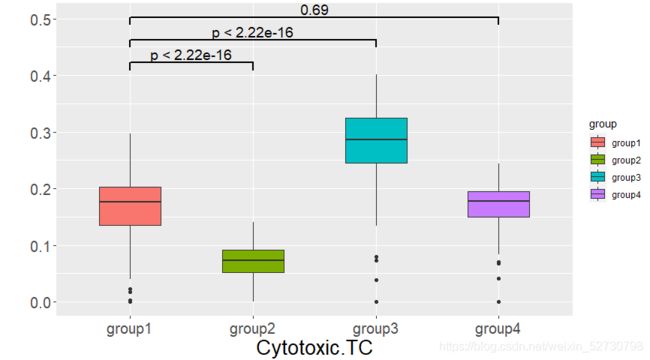
代码如下:
library("ggsignif")
library(gridExtra)
library(tibble)#rownames_to_column
library(ggplot2)
library(grid)
library(reshape2)
library(ggpubr)
library(dplyr)
library(tidyr)
library(pheatmap)
pkgs <- c("matrixStats", "pheatmap", "RColorBrewer", "tidyverse", "cowplot","ggpubr","bslib","ggthemes")
lapply(pkgs, library, character.only = T)#Error in theme_base() : 没有"theme_base"这个函数
plot.info <- read.table('ImmuCellAI_icb_result.txt',
sep = '\t',
header = T,
row.names = 1)
plot.info <- plot.info[,-2]###去除respond列
#plot.info <- plot.info[,-17]###去除数据太低的Effect-memory,如果对数据有改动请注意修改
plot.info <- plot.info[,-25]###去除gamma
plot.info <- plot.info[,-25]###去除score
###################################################
plot.info_1 <- plot.info %>%
as.data.frame() %>%
rownames_to_column("sample") %>%
pivot_longer(cols = 3,
names_to = "CellType",
values_to = "Composition")
###分组进行绘图
plot.info_1 <- plot.info_1[,c(25,2,26)]
plot1 <- ggboxplot(
plot.info_1,
x = "CellType",
y = "Composition",
color = "black",
fill = "group", #只需要修改这里,讲fill = "CellType"改为fill = "group"
xlab = "",
ylab = "",
#main = "TC",
col="white",
borde="white"
)
###分组并计算差异进行绘图
geom_signif()
compare_means(Composition ~ group, #将Composition与group进行联系
data = plot.info_1,
method="wilcox.test",
paired=FALSE)#计算Wilcoxon
my_comparisons <- list(c("group1","group2"), c("group1","group3"),c("group1", "group4"))#这里设置对照组
ggplot(plot.info_1,aes(group,
Composition,
fill=group#CellType#设置
))+
geom_boxplot(width=0.5)+
theme(plot.title=element_text(size = 10),
axis.text.x=element_text(size=15,angle=0),
axis.text.y=element_text(size=15),
axis.title.x=element_text(size = 20),
axis.title.y=element_text(size = 25))+
labs(x="Cytotoxic.TC", #设置CellType#横坐标显示内容,可以为空
y= "" #纵坐标显示内容,可以为空
)+
geom_signif(comparisons = my_comparisons,step_increase = 0.1,#两组直接对比框的高度
map_signif_level = F,#这里可以设置两组之间差异显示为数值或者***,F为数值,T为***
test = t.test,size=1,#线条粗细
textsize = 5#P值字号大小
)此处代码仅供参考
我把代码与数据文件上传了CSDN资源
https://download.csdn.net/download/weixin_52730798/20336446?spm=1001.2014.3001.5503
有兴趣的可以支持一下