前情回顾
Seurat 4.0 ||单细胞数据分析工具箱有更新
Seurat 4.0 ||单细胞多模态数据整合算法WNN
Seurat 4.0 || 分析scRNA和表面抗体数据
单细胞数据数据正在走向统一:在一个细胞内同时测量分属中心法则不同阶段的多种数据类型。这需要我们开发新的方法来将这些数据整合在一起以描绘一个完整的细胞状态。Seurat升级到4.0以后,在一个Seurat对象中可以存储(数据结构)和计算(算法)单细胞多模态数据。本文我们跟着官方教程演示使用WNN分析多模态技术分析10xscRNA+ATAC数据。使用的数据集在10x网站上公开,是为10412个同时测量转录组和ATAC的PBMCs细胞。
在这个例子中,我们将演示:
- 使用成对的转录组和ATAC-seq数据创建多模式Seurat对象
- 对单细胞RNA+ATAC数据进行加权近邻聚类
- 利用这两种模式来识别不同细胞类型和状态的调节因子
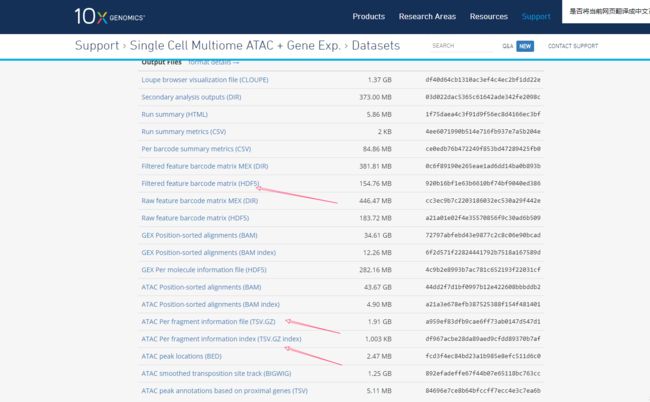
你可以从10x dataset网站下载数据集。请务必下载以下文件:
Filtered feature barcode matrix (HDF5)
ATAC Per fragment information file (TSV.GZ)
ATAC Per fragment information index (TSV.GZ index) # 这个索引文件别忘了哦
网址:https://support.10xgenomics.com/single-cell-multiome-atac-gex/datasets/1.0.0/pbmc_granulocyte_sorted_10k?
最后,为了运行本示例,请确保安装了以下软件包:
- Seurat v4.
- Signac for the analysis of single-cell chromatin datasets
- EnsDb.Hsapiens.v86 for a set of annotations for hg38
- dplyr to help manipulate data tables
library(Seurat)
library(Signac)
library(EnsDb.Hsapiens.v86)
library(dplyr)
library(ggplot2)
我们将基于基因表达数据创建一个Seurat对象,然后添加ATAC-seq数据作为第二个assay。关于创建和处理ATAC对象的更多信息,可以浏览Signac相关文档。
# the 10x hdf5 file contains both data types.
inputdata.10x <- Read10X_h5("../data/pbmc_granulocyte_sorted_10k_filtered_feature_bc_matrix.h5")
# extract RNA and ATAC data
rna_counts <- inputdata.10x$`Gene Expression`
atac_counts <- inputdata.10x$Peaks
# Create Seurat object
pbmc <- CreateSeuratObject(counts = rna_counts)
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
head([email protected])
orig.ident nCount_RNA nFeature_RNA percent.mt
AAACAGCCAAGGAATC-1 SeuratProject 8380 3308 7.470167
AAACAGCCAATCCCTT-1 SeuratProject 3771 1896 10.527711
AAACAGCCAATGCGCT-1 SeuratProject 6876 2904 6.457243
AAACAGCCACACTAAT-1 SeuratProject 1733 846 18.003462
AAACAGCCACCAACCG-1 SeuratProject 5415 2282 6.500462
AAACAGCCAGGATAAC-1 SeuratProject 2759 1353 6.922798
VlnPlot(pbmc,features=c('percent.mt','nCount_RNA', 'nFeature_RNA'),pt.size = 0,cols = "red")

添加ATAC数据。
# Now add in the ATAC-seq data
# we'll only use peaks in standard chromosomes
grange.counts <- StringToGRanges(rownames(atac_counts), sep = c(":", "-"))
grange.use <- seqnames(grange.counts) %in% standardChromosomes(grange.counts)
atac_counts <- atac_counts[as.vector(grange.use), ]
annotations <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v86)
seqlevelsStyle(annotations) <- 'UCSC'
genome(annotations) <- "hg38"
frag.file <- "/home/data/vip06/dataset/pbmc_granulocyte_sorted_10k_atac_fragments.tsv.gz"
chrom_assay <- CreateChromatinAssay(
counts = atac_counts,
sep = c(":", "-"),
genome = 'hg38',
fragments = frag.file,
min.cells = 10,
annotation = annotations
)
pbmc[["ATAC"]] <- chrom_assay
查看数据集对象:
pbmc[["ATAC"]]
ChromatinAssay data with 105913 features for 11909 cells
Variable features: 0
Genome: hg38
Annotation present: TRUE
Motifs present: FALSE
Fragment files: 1
pbmc
An object of class Seurat
142514 features across 11909 samples within 2 assays
Active assay: RNA (36601 features, 0 variable features)
1 other assay present: ATAC
我们根据每种模式检测到的分子数以及线粒体百分比来执行基本的QC,其实这一步什么也没有Q掉,只是获得数据概览,了解数据的质量分布。
head([email protected])
orig.ident nCount_RNA nFeature_RNA percent.mt nCount_ATAC nFeature_ATAC
AAACAGCCAAGGAATC-1 SeuratProject 8380 3308 7.470167 55550 13867
AAACAGCCAATCCCTT-1 SeuratProject 3771 1896 10.527711 20485 7247
AAACAGCCAATGCGCT-1 SeuratProject 6876 2904 6.457243 16674 6528
AAACAGCCACACTAAT-1 SeuratProject 1733 846 18.003462 2007 906
AAACAGCCACCAACCG-1 SeuratProject 5415 2282 6.500462 7658 3323
AAACAGCCAGGATAAC-1 SeuratProject 2759 1353 6.922798 10355 4267
VlnPlot(pbmc, features = c("nCount_RNA", "nFeature_RNA", "percent.mt", "nCount_ATAC", "nFeature_ATAC"), ncol = 3,
log = TRUE, pt.size = 0) + NoLegend()

如果是多个样本这样的展示方式可以看到每个样本的质量差异,咦,WNN框架下如何执行多样本分析?这里我们还是假设有那么一组吧,人为设置一个分组。
pbmc$replicate <- sample(c("ctrl", "para"), size = ncol(pbmc), replace = TRUE)
VlnPlot(pbmc, features = c("nCount_RNA", "nFeature_RNA", "percent.mt", "nCount_ATAC", "nFeature_ATAC"), ncol = 3,group.by = 'replicate',
log = TRUE, pt.size = 0) + NoLegend()

看到数据的质量分布之后,我们可以执行真正的质控了,把异常值或者不是细胞的barcode去掉,保证进入分析的都是细胞,而非杂质。
pbmc <- subset(
x = pbmc,
subset = nCount_ATAC < 7e4 &
nCount_ATAC > 5e3 &
nCount_RNA < 25000 &
nCount_RNA > 1000 &
percent.mt < 20
)
接下来,我们使用RNA和ATAC-seq数据的标准方法,分别对这两种检测方法进行预处理和降维。
# RNA analysis
DefaultAssay(pbmc) <- "RNA"
pbmc <- SCTransform(pbmc, verbose = FALSE) %>% RunPCA() %>% RunUMAP(dims = 1:50, reduction.name = 'umap.rna', reduction.key = 'rnaUMAP_')
PC_ 1
Positive: VCAN, PLXDC2, SAT1, SLC8A1, NAMPT, NEAT1, ZEB2, DPYD, LRMDA, LYZ
FCN1, LYN, ANXA1, JAK2, CD36, AOAH, ARHGAP26, TYMP, RBM47, PLCB1
PID1, LRRK2, ACSL1, LYST, IRAK3, DMXL2, MCTP1, AC020916.1, GAB2, TBXAS1
Negative: EEF1A1, RPL13, BCL2, RPS27, RPL13A, LEF1, BCL11B, BACH2, INPP4B, RPL41
IL32, RPS27A, CAMK4, IL7R, RPL3, SKAP1, RPS2, GNLY, LTB, CD247
RPS12, RPS18, RPL11, PRKCH, ANK3, RPL23A, THEMIS, CCL5, RPL10, RPS3
PC_ 2
Positive: BANK1, IGHM, AFF3, IGKC, RALGPS2, MS4A1, PAX5, CD74, EBF1, FCRL1
CD79A, OSBPL10, LINC00926, COL19A1, BLK, NIBAN3, IGHD, CD22, HLA-DRA, ADAM28
CD79B, PLEKHG1, COBLL1, AP002075.1, LIX1-AS1, CCSER1, TCF4, BCL11A, GNG7, STEAP1B
Negative: GNLY, CCL5, NKG7, CD247, IL32, PRKCH, PRF1, BCL11B, INPP4B, LEF1
IL7R, CAMK4, THEMIS, GZMA, RORA, GZMH, TXK, TC2N, TRBC1, PITPNC1
SYNE2, PDE3B, STAT4, TRAC, TGFBR3, VCAN, KLRD1, NCALD, CTSW, SLFN12L
PC_ 3
Positive: LEF1, CAMK4, INPP4B, PDE3B, IL7R, FHIT, BACH2, MAML2, TSHZ2, SERINC5
BCL2, EEF1A1, NELL2, ANK3, CCR7, PRKCA, TCF7, BCL11B, LTB, AC139720.1
OXNAD1, MLLT3, RPL11, AL589693.1, TRABD2A, RPL13, RASGRF2, MAL, NR3C2, LDHB
Negative: GNLY, NKG7, CCL5, PRF1, GZMA, GZMH, KLRD1, SPON2, FGFBP2, CST7
GZMB, CCL4, TGFBR3, KLRF1, CTSW, BNC2, ADGRG1, PDGFD, PPP2R2B, PYHIN1
IL2RB, CLIC3, A2M, NCAM1, MCTP2, TRDC, C1orf21, SAMD3, MYBL1, FCRL6
PC_ 4
Positive: TCF4, CUX2, AC023590.1, LINC01374, RHEX, LINC01478, EPHB1, PLD4, PTPRS, ZFAT
LINC00996, CLEC4C, LILRA4, COL26A1, PLXNA4, SCN9A, CCDC50, FAM160A1, ITM2C, COL24A1
UGCG, RUNX2, GZMB, NRP1, PHEX, IRF8, JCHAIN, SLC35F3, PACSIN1, AC007381.1
Negative: BANK1, IGHM, MS4A1, PAX5, IGKC, FCRL1, EBF1, RALGPS2, CD79A, LINC00926
OSBPL10, GNLY, COL19A1, IGHD, NKG7, CD22, BACH2, CCL5, PLEKHG1, CD79B
ADAM28, LIX1-AS1, LARGE1, STEAP1B, AP002075.1, AC120193.1, BLK, FCER2, ARHGAP24, IFNG-AS1
PC_ 5
Positive: VCAN, PLCB1, ANXA1, PLXDC2, DPYD, CD36, ARHGAP26, ARHGAP24, ACSL1, AC020916.1
LRMDA, DYSF, CSF3R, MEGF9, PDE4D, S100A8, FNDC3B, ADAMTSL4-AS1, S100A9, TMTC2
RAB11FIP1, CREB5, LRRK2, NAMPT, RBM47, JUN, SLC2A3, GNLY, GLT1D1, VCAN-AS1
Negative: CDKN1C, FCGR3A, TCF7L2, IFITM3, CST3, MTSS1, AIF1, LST1, MS4A7, PSAP
WARS, ACTB, HLA-DPA1, SERPINA1, CD74, FCER1G, IFI30, COTL1, CFD, SMIM25
HLA-DRA, HLA-DRB1, FMNL2, CSF1R, FTL, MAFB, TMSB4X, TYROBP, HLA-DPB1, CCDC26
06:10:45 UMAP embedding parameters a = 0.9922 b = 1.112
06:10:45 Read 10412 rows and found 50 numeric columns
06:10:45 Using Annoy for neighbor search, n_neighbors = 30
06:10:45 Building Annoy index with metric = cosine, n_trees = 50
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
06:10:46 Writing NN index file to temp file /tmp/Rtmp3eAPNF/file3dd9069c738d7
06:10:46 Searching Annoy index using 1 thread, search_k = 3000
06:10:49 Annoy recall = 100%
06:10:50 Commencing smooth kNN distance calibration using 1 thread
06:10:52 Initializing from normalized Laplacian + noise
06:10:53 Commencing optimization for 200 epochs, with 443480 positive edges
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
06:10:59 Optimization finished
There were 50 or more warnings (use warnings() to see the first 50)
分析ATAC数据,ATAC数据在这里也是一个矩阵,只是不是RNA含量而是fragments矩阵。但是ATAC的assays更丰富。
str(pbmc@assays$ATAC,max.level = 2)
Formal class 'ChromatinAssay' [package "Signac"] with 16 slots
..@ ranges :Formal class 'GRanges' [package "GenomicRanges"] with 7 slots
..@ motifs : NULL
..@ fragments :List of 1
..@ seqinfo :Formal class 'Seqinfo' [package "GenomeInfoDb"] with 4 slots
..@ annotation :Formal class 'GRanges' [package "GenomicRanges"] with 7 slots
..@ bias : NULL
..@ positionEnrichment: list()
..@ links :Formal class 'GRanges' [package "GenomicRanges"] with 7 slots
..@ counts :Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
..@ data :Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
..@ scale.data : num[0 , 0 ]
..@ key : chr "atac_"
..@ assay.orig : NULL
..@ var.features : chr [1:105913] "chr19-1230013-1281741" "chr19-39381822-39438328" "chr17-7832492-7859042" "chr19-12774552-12797715" ...
..@ meta.features :'data.frame': 105913 obs. of 2 variables:
..@ misc : NULL
如fragments矩阵信息
head(pbmc@assays$ATAC@counts)
6 x 10412 sparse Matrix of class "dgCMatrix"
[[ suppressing 76 column names 'AAACAGCCAAGGAATC-1', 'AAACAGCCAATCCCTT-1', 'AAACAGCCAATGCGCT-1' ... ]]
chr1-10109-10357 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
chr1-180730-181630 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
chr1-191491-191736 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
chr1-267816-268196 . . . . . . . . . . . . . . . 2 . . . . . . . . . . 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
chr1-586028-586373 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
chr1-629721-630172 . . . . . . . . . . . . . 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
chr1-10109-10357 . . . ......
chr1-180730-181630 . . . ......
chr1-191491-191736 . . . ......
chr1-267816-268196 . . . ......
chr1-586028-586373 . . . ......
chr1-629721-630172 . . . ......
.....suppressing 10336 columns in show(); maybe adjust 'options(max.print= *, width = *)'
..............................
如注释信息:
head(pbmc@assays$ATAC@annotation)
GRanges object with 6 ranges and 5 metadata columns:
seqnames ranges strand | tx_id gene_name gene_id gene_biotype type
|
ENSE00001489430 chrX 276322-276394 + | ENST00000399012 PLCXD1 ENSG00000182378 protein_coding exon
ENSE00001536003 chrX 276324-276394 + | ENST00000484611 PLCXD1 ENSG00000182378 protein_coding exon
ENSE00002160563 chrX 276353-276394 + | ENST00000430923 PLCXD1 ENSG00000182378 protein_coding exon
ENSE00001750899 chrX 281055-281121 + | ENST00000445062 PLCXD1 ENSG00000182378 protein_coding exon
ENSE00001489388 chrX 281192-281684 + | ENST00000381657 PLCXD1 ENSG00000182378 protein_coding exon
ENSE00001719251 chrX 281194-281256 + | ENST00000429181 PLCXD1 ENSG00000182378 protein_coding exon
-------
seqinfo: 25 sequences from hg38 genome
理解Seurat的数据是很重要的,不然很多分析数据可能不知道如何提取调用,也不知道函数做了什么。在这之后,我们进行ATAC数据的标准分析。
# ATAC analysis
# We exclude the first dimension as this is typically correlated with sequencing depth
DefaultAssay(pbmc) <- "ATAC"
pbmc <- RunTFIDF(pbmc)
pbmc <- FindTopFeatures(pbmc, min.cutoff = 'q0')
pbmc <- RunSVD(pbmc)
pbmc <- RunUMAP(pbmc, reduction = 'lsi', dims = 2:50, reduction.name = "umap.atac", reduction.key = "atacUMAP_")
注意,这里的以及所有特征选择之后的再次降维用到的维度dims,都需要谨慎选择,我们知道每个坐标值都有一系列特征支持程度(如loading值),可以反映出该维度代表的生物学意义。如这里的dims = 2:50,排除了第一个维度,因为作者发现它通常与排序深度相关
pbmc@[email protected][1:4,1:4]
LSI_1 LSI_2 LSI_3 LSI_4
chr19-1230013-1281741 0.008759956 0.0022949972 -0.0008663089 -0.0019122743
chr19-39381822-39438328 0.008735519 -0.0007909501 -0.0018568479 -0.0018096328
chr17-7832492-7859042 0.008713235 0.0012451020 -0.0006112821 0.0004232284
chr19-12774552-12797715 0.008700547 0.0015833131 -0.0014307690 0.0013240653
用Signac分析过scATAC的同学对这里的流程不会感到陌生,如果是第一次接触ATAC分析会觉得比较陌生了,如RunTFIDF是什么?这时候我们可以?RunTFIDF来查看具体信息以及参考文献。
?RunTFIDF
method
Which TF-IDF implementation to use. Choice of:
* 1: The TF-IDF implementation used by Stuart & Butler et al. 2019 (https://doi.org/10.1101/460147). This computes *\log(TF \times IDF)*.
* 2: The TF-IDF implementation used by Cusanovich & Hill et al. 2018 (https://doi.org/10.1016/j.cell.2018.06.052). This computes *TF \times (\log(IDF))*
* 3: The log-TF method used by Andrew Hill (http://andrewjohnhill.com/blog/2019/05/06/dimensionality-reduction-for-scatac-data/). This computes *\log(TF) \times \log(IDF)*.
* 4: The 10x Genomics method (no TF normalization). This computes *IDF*.

我们计算一个WNN图,表示RNA和ATAC-seq模式的加权组合。我们将此图用于UMAP的可视化和聚类。我们在Seurat 4.0 || 分析scRNA和表面抗体数据
中查看过FindMultiModalNeighbors之后的数据结构。
pbmc <- FindMultiModalNeighbors(pbmc, reduction.list = list("pca", "lsi"), dims.list = list(1:50, 2:50))
Calculating cell-specific modality weights
Finding 20 nearest neighbors for each modality.
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=05s
Calculating kernel bandwidths
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=01s
Finding multimodal neighbors
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=20s
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=01s
Constructing multimodal KNN graph
Constructing multimodal SNN graph
pbmc <- RunUMAP(pbmc, nn.name = "weighted.nn", reduction.name = "wnn.umap", reduction.key = "wnnUMAP_")
06:41:32 UMAP embedding parameters a = 0.9922 b = 1.112
06:41:33 Commencing smooth kNN distance calibration using 1 thread
06:41:35 Initializing from normalized Laplacian + noise
06:41:36 Commencing optimization for 200 epochs, with 308698 positive edges
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
06:41:42 Optimization finished
pbmc <- FindClusters(pbmc, graph.name = "wsnn", algorithm = 3, verbose = FALSE)
这里聚类用的算法是SLM
algorithm
Algorithm for modularity optimization
(1 = original Louvain algorithm;
2 = Louvain algorithm with multilevel refinement;
3 = SLM algorithm;
4 = Leiden algorithm). Leiden requires the leidenalg python.
这些事图聚类的话语体系,我们知道在构建了细胞间的图结构之后,要聚类就需要计算细胞间的相互关系,这里是网略数据科学的一个重要领域:社区发现(community detection),如这里的SLM (smart local moving)算法是一种在大型网络中进行社区发现(或聚类)的算法。SLM算法最大化了所谓的模块化函数。该算法已成功应用于具有数千万个节点和数亿条边的网络。算法的细节记录在一篇论文中(Waltman & Van Eck, 2013)。在igraph中实现SLM可以这样:
#devtools::install_github("chen198328/slm")
library(igraph)
library(slm)
net <- graph_from_adjacency_matrix(pbmc[["wsnn"]])
slm.net <-slm.community(net)
当然这对大部分不做算法调整的同学来讲并没有什么用,这里只是说明我们的聚类是在图空间中进行的。
接下来我们对细胞群进行注释。注意,您还可以使用我们构建好的PBMC参考数据集来进行注释,可以用Seurat(vignette见官网)或自动化web工具:Azimuth。
# perform sub-clustering on cluster 6 to find additional structure
pbmc <- FindSubCluster(pbmc, cluster = 6, graph.name = "wsnn", algorithm = 3)
Idents(pbmc) <- "sub.cluster"
指定某群在聚类这个函数好用啊
table(pbmc$seurat_clusters)
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
2386 1159 973 606 558 502 498 470 438 438 372 352 329 285 256 198 163 142 137 106 26 18
> table(pbmc$sub.cluster)
0 1 10 11 12 13 14 15 16 17 18 19 2 20 21 3 4 5 6_0 6_1 7 8 9
2386 1159 372 352 329 285 256 198 163 142 137 106 973 26 18 606 558 502 274 224 470 438 438
其他没变,cluster被分成了两组:6_0 ,6_1。
# add annotations
pbmc <- RenameIdents(pbmc, '19' = 'pDC','20' = 'HSPC','15' = 'cDC')
pbmc <- RenameIdents(pbmc, '0' = 'CD14 Mono', '8' ='CD14 Mono', '5' = 'CD16 Mono')
pbmc <- RenameIdents(pbmc, '17' = 'Naive B', '11' = 'Intermediate B', '10' = 'Memory B', '21' = 'Plasma')
pbmc <- RenameIdents(pbmc, '7' = 'NK')
pbmc <- RenameIdents(pbmc, '4' = 'CD4 TCM', '13'= "CD4 TEM", '3' = "CD4 TCM", '16' ="Treg", '1' ="CD4 Naive", '14' = "CD4 Naive")
pbmc <- RenameIdents(pbmc, '2' = 'CD8 Naive', '9'= "CD8 Naive", '12' = 'CD8 TEM_1', '6_0' = 'CD8 TEM_2', '6_1' ='CD8 TEM_2')
pbmc <- RenameIdents(pbmc, '18' = 'MAIT')
pbmc <- RenameIdents(pbmc, '6_2' ='gdT', '6_3' = 'gdT')
pbmc$celltype <- Idents(pbmc)
pbmc
An object of class Seurat
166710 features across 10412 samples within 3 assays
Active assay: ATAC (105913 features, 105913 variable features)
2 other assays present: RNA, SCT
5 dimensional reductions calculated: pca, umap.rna, lsi, umap.atac, wnn.umap
head([email protected])
orig.ident nCount_RNA nFeature_RNA percent.mt nCount_ATAC nFeature_ATAC replicate nCount_SCT nFeature_SCT SCT.weight wsnn_res.0.8
AAACAGCCAAGGAATC-1 SeuratProject 8380 3308 7.470167 55550 13867 para 4685 2693 0.4514450 1
AAACAGCCAATCCCTT-1 SeuratProject 3771 1896 10.527711 20485 7247 para 3781 1895 0.5069799 4
AAACAGCCAATGCGCT-1 SeuratProject 6876 2904 6.457243 16674 6528 para 4686 2769 0.4324867 1
AAACAGCCACCAACCG-1 SeuratProject 5415 2282 6.500462 7658 3323 ctrl 4486 2278 0.4738122 2
AAACAGCCAGGATAAC-1 SeuratProject 2759 1353 6.922798 10355 4267 ctrl 3332 1352 0.5361155 1
AAACAGCCAGTAGGTG-1 SeuratProject 7614 3061 6.895193 39441 11628 ctrl 4711 2764 0.5273038 9
seurat_clusters sub.cluster celltype
AAACAGCCAAGGAATC-1 1 1 CD4 Naive
AAACAGCCAATCCCTT-1 4 4 CD4 TCM
AAACAGCCAATGCGCT-1 1 1 CD4 Naive
AAACAGCCACCAACCG-1 2 2 CD8 Naive
AAACAGCCAGGATAAC-1 1 1 CD4 Naive
AAACAGCCAGTAGGTG-1 9 9 CD8 Naive
基于基因表达可视化聚类,ATAC-seq,或WNN分析。与前面的分析相比,差异更加微妙(您可以探索权重,它比我们的CITE-seq示例中更均匀地分割),但是我们发现WNN提供了最清晰的细胞状态分离。
p1 <- DimPlot(pbmc, reduction = "umap.rna", group.by = "celltype", label = TRUE, label.size = 2.5, repel = TRUE) + ggtitle("RNA")
p2 <- DimPlot(pbmc, reduction = "umap.atac", group.by = "celltype", label = TRUE, label.size = 2.5, repel = TRUE) + ggtitle("ATAC")
p3 <- DimPlot(pbmc, reduction = "wnn.umap", group.by = "celltype", label = TRUE, label.size = 2.5, repel = TRUE) + ggtitle("WNN")
p1 + p2 + p3 & NoLegend() & theme(plot.title = element_text(hjust = 0.5))
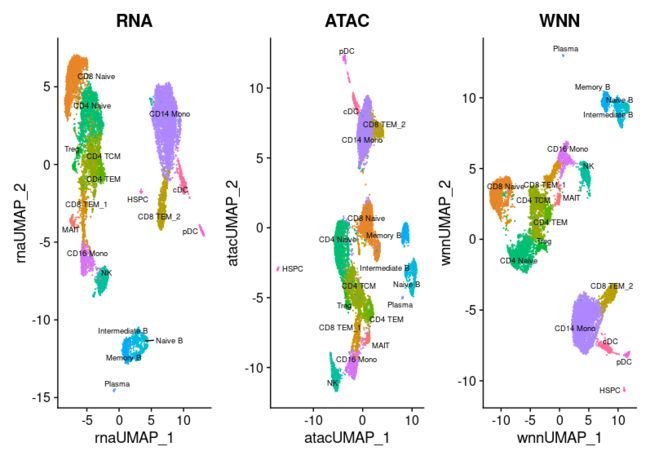
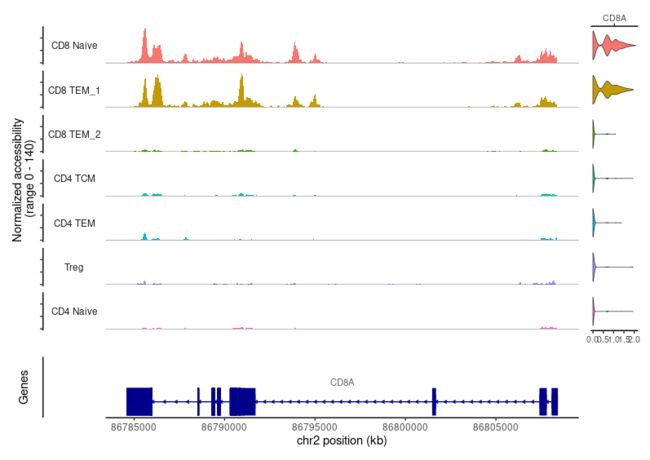
例如,ATAC-seq数据有助于CD4和CD8 T细胞状态的分离,这是由于多个基因座的存在。在不同的T细胞亚型之间表现出不同的可及性。例如,我们可以利用Signac可视化工具,将CD8A位点的“伪体(pseudobulk)”轨迹与基因表达水平的小提琴图一起可视化。
## to make the visualization easier, subset T cell clusters
celltype.names <- levels(pbmc)
tcell.names <- grep("CD4|CD8|Treg", celltype.names,value = TRUE)
tcells <- subset(pbmc, idents = tcell.names)
CoveragePlot(tcells, region = 'CD8A', features = 'CD8A', assay = 'ATAC', expression.assay = 'SCT', peaks = FALSE)
接下来,我们将检查每个细胞的可达区域,以确定富集基序(enriched motifs)。正如Signac motifs vignette中所描述的,有几种方法可以做到这一点,但我们将使用Greenleaf实验室的chromVAR包。这将计算已知motifs的每个单元的可访问性得分,并将这些得分作为Seurat对象中的第三个分析(chromVAR)添加。
再走下去,请确保安装了以下软件包。
- chromVAR for the analysis of motif accessibility in scATAC-seq
- presto for fast differential expression analyses.
- TFBSTools for TFBS analysis
- JASPAR2020 for JASPAR motif models
- motifmatchr for motif matching
- BSgenome.Hsapiens.UCSC.hg38 for chromVAR
我先library一下看看哪些是安装过的。
library(chromVAR)
library(JASPAR2020)
library(TFBSTools)
library(motifmatchr)
library(BSgenome.Hsapiens.UCSC.hg38)
针对没安装的,我们可以这样来安装。
remotes::install_github("immunogenomics/presto")
BiocManager::install(c("chromVAR", "TFBSTools", "JASPAR2020", "motifmatchr", "BSgenome.Hsapiens.UCSC.hg38"))
# Scan the DNA sequence of each peak for the presence of each motif, and create a Motif object
DefaultAssay(pbmc) <- "ATAC"
pwm_set <- getMatrixSet(x = JASPAR2020, opts = list(species = 9606, all_versions = FALSE))
motif.matrix <- CreateMotifMatrix(features = granges(pbmc), pwm = pwm_set, genome = 'hg38', use.counts = FALSE)
motif.object <- CreateMotifObject(data = motif.matrix, pwm = pwm_set)
pbmc <- SetAssayData(pbmc, assay = 'ATAC', slot = 'motifs', new.data = motif.object)
# Note that this step can take 30-60 minutes
pbmc <- RunChromVAR(
object = pbmc,
genome = BSgenome.Hsapiens.UCSC.hg38
)
pbmc
An object of class Seurat
166710 features across 10412 samples within 3 assays
Active assay: ATAC (105913 features, 105913 variable features)
2 other assays present: RNA, SCT
5 dimensional reductions calculated: pca, umap.rna, lsi, umap.atac, wnn.umap
最后,我们探索多模态数据集来识别每个细胞状态的关键调节器(key regulators)。配对数据提供了一个独特的机会来识别满足多种标准的转录因子(TFs),帮助缩小假定的调控因子的名单列表。我们的目标是在RNA测量中识别在多种细胞类型中表达丰富的TFs,同时在ATAC测量中富集其motifs 的可达性。
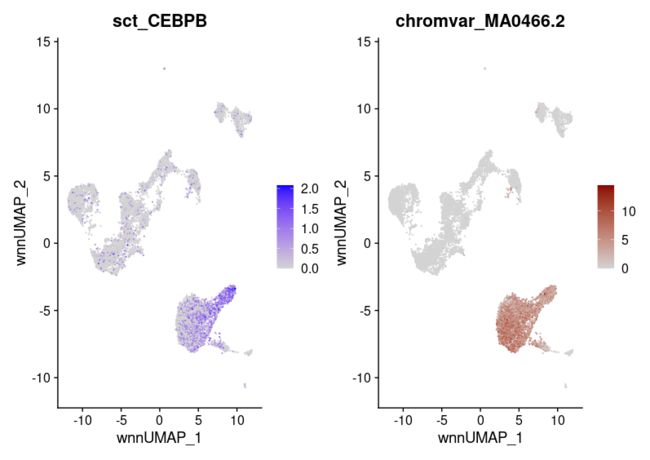
作为阳性对照,CCAAT增强子结合蛋白(CEBP)家族蛋白,包括TF CEBPB,已多次证明在单核细胞和树突状细胞等髓系细胞的分化和功能中发挥重要作用。我们可以看到CEBPB的表达和编码CEBPB结合位点的MA0466.2.4基序的可及性都在单核细胞中富集。
#returns MA0466.2
motif.name <- ConvertMotifID(pbmc, name = 'CEBPB')
gene_plot <- FeaturePlot(pbmc, features = "sct_CEBPB", reduction = 'wnn.umap')
motif_plot <- FeaturePlot(pbmc, features = motif.name, min.cutoff = 0, cols = c("lightgrey", "darkred"), reduction = 'wnn.umap')
gene_plot | motif_plot
我们想要量化这个关系,并搜索所有的细胞类型来找到类似的例子。为此,我们将使用presto包来执行快差异分析。我们进行了两项测试 :一项使用基因表达数据,另一项使用chromVAR motif可达性。presto根据Wilcox秩和检验(这也是Seurat的默认方法)计算一个p值,我们将搜索限制为在两个测试中都返回显著结果的TFs。
presto还计算了“AUC”统计值,它反映了每个基因(或基序)作为细胞类型标记的能力。曲线下面积(AUC)的最大值为1表示标记良好。由于基因和基序的AUC统计值是相同的,因此我们将两种测试的AUC值取平均值,并以此来对每种细胞类型的TFs进行排序:
markers_rna <- presto:::wilcoxauc.Seurat(X = pbmc, group_by = 'celltype', assay = 'data', seurat_assay = 'SCT')
markers_motifs <- presto:::wilcoxauc.Seurat(X = pbmc, group_by = 'celltype', assay = 'data', seurat_assay = 'chromvar')
motif.names <- markers_motifs$feature
colnames(markers_rna) <- paste0("RNA.", colnames(markers_rna))
colnames(markers_motifs) <- paste0("motif.", colnames(markers_motifs))
markers_rna$gene <- markers_rna$RNA.feature
markers_motifs$gene <- ConvertMotifID(pbmc, id = motif.names)
head(markers_motifs)
motif.feature motif.group motif.avgExpr motif.logFC motif.statistic motif.auc motif.pval motif.padj motif.pct_in
1 MA0030.1 CD14 Mono -0.2070450 -0.2837042 8748968 0.4082863 4.331768e-47 5.397656e-47 100
2 MA0031.1 CD14 Mono -0.6256700 -0.8643914 5368273 0.2505201 0.000000e+00 0.000000e+00 100
3 MA0051.1 CD14 Mono 0.1237673 0.1974597 11955125 0.5579074 9.048795e-20 1.045235e-19 100
4 MA0057.1 CD14 Mono 0.7245745 1.0173693 16968911 0.7918847 0.000000e+00 0.000000e+00 100
5 MA0059.1 CD14 Mono 0.9826472 1.3781270 17525266 0.8178480 0.000000e+00 0.000000e+00 100
6 MA0066.1 CD14 Mono 1.6772753 2.3573633 19347910 0.9029049 0.000000e+00 0.000000e+00 100
motif.pct_out gene
1 100 FOXF2
2 100 FOXD1
3 100 IRF2
4 100 MZF1(var.2)
5 100 MAX::MYC
6 100 PPARG
寻找toptf的函数
# a simple function to implement the procedure above
topTFs <- function(celltype, padj.cutoff = 1e-2) {
ctmarkers_rna <- dplyr::filter(
markers_rna, RNA.group == celltype, RNA.padj < padj.cutoff, RNA.logFC > 0) %>%
arrange(-RNA.auc)
ctmarkers_motif <- dplyr::filter(
markers_motifs, motif.group == celltype, motif.padj < padj.cutoff, motif.logFC > 0) %>%
arrange(-motif.auc)
top_tfs <- inner_join(
x = ctmarkers_rna[, c(2, 11, 6, 7)],
y = ctmarkers_motif[, c(2, 1, 11, 6, 7)], by = "gene"
)
top_tfs$avg_auc <- (top_tfs$RNA.auc + top_tfs$motif.auc) / 2
top_tfs <- arrange(top_tfs, -avg_auc)
return(top_tfs)
}
我们现在可以计算和可视化任何细胞类型的putative regulators。我们恢复了成熟的调控因子,包括用于NK细胞的TBX21(这里的背景知识很重要啊),浆细胞的IRF4,造血祖细胞的SOX4,B细胞的EBF1和PAX5,pDC的IRF8和TCF4。我们相信,类似的策略可以用来帮助关注不同系统中一组putative regulators。

# identify top markers in NK and visualize
head(topTFs("NK"), 3)
head(topTFs("NK"), 3)
RNA.group gene RNA.auc RNA.pval motif.group motif.feature motif.auc motif.pval avg_auc
1 NK TBX21 0.7269867 0.000000e+00 NK MA0690.1 0.9175550 3.489416e-206 0.8222709
2 NK EOMES 0.5898072 8.133395e-101 NK MA0800.1 0.9238892 2.013461e-212 0.7568482
3 NK RUNX3 0.7712620 9.023079e-121 NK MA0684.2 0.6923503 3.069778e-45 0.7318062
motif.name <- ConvertMotifID(pbmc, name = 'TBX21')
gene_plot <- FeaturePlot(pbmc, features = "sct_TBX21", reduction = 'wnn.umap')
motif_plot <- FeaturePlot(pbmc, features = motif.name, min.cutoff = 0, cols = c("lightgrey", "darkred"), reduction = 'wnn.umap')
gene_plot | motif_plot

# identify top markers in pDC and visualize
head(topTFs("pDC"), 3)
head(topTFs("pDC"), 3)
RNA.group gene RNA.auc RNA.pval motif.group motif.feature motif.auc motif.pval avg_auc
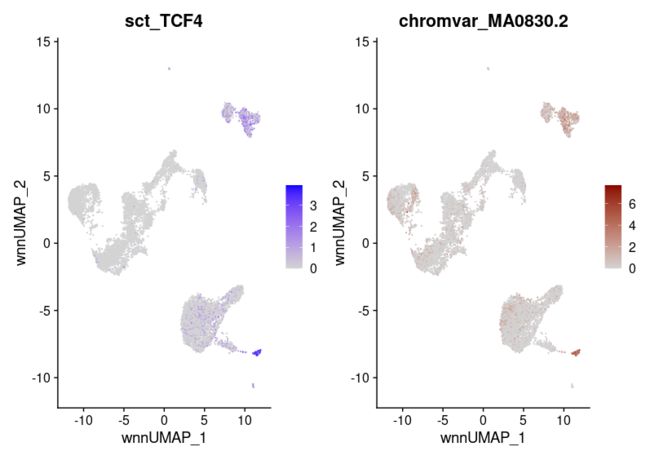
1 pDC TCF4 0.9998833 3.347777e-163 pDC MA0830.2 0.9952958 3.912770e-69 0.9975896
2 pDC IRF8 0.9905372 2.063258e-124 pDC MA0652.1 0.8714643 1.143055e-39 0.9310008
3 pDC RUNX2 0.9754498 7.815119e-112 pDC MA0511.2 0.8355034 1.126551e-32 0.9054766
motif.name <- ConvertMotifID(pbmc, name = 'TCF4')
gene_plot <- FeaturePlot(pbmc, features = "sct_TCF4", reduction = 'wnn.umap')
motif_plot <- FeaturePlot(pbmc, features = motif.name, min.cutoff = 0, cols = c("lightgrey", "darkred"), reduction = 'wnn.umap')
gene_plot | motif_plot
# identify top markers in HSPC and visualize
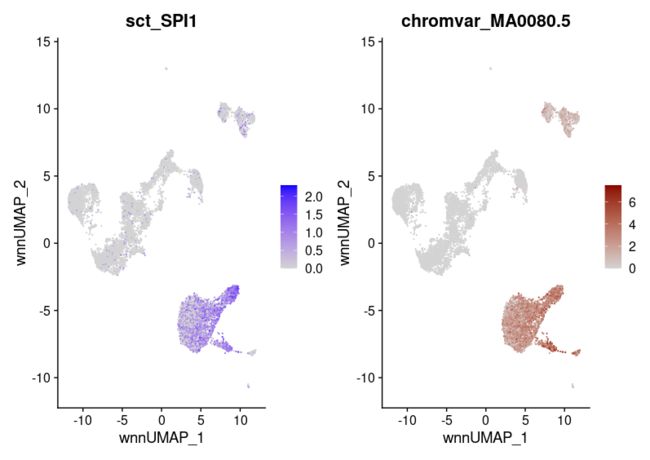
head(topTFs("CD16 Mono"),3)
RNA.group gene RNA.auc RNA.pval motif.group motif.feature motif.auc motif.pval avg_auc
1 CD16 Mono TBX21 0.6550599 8.750032e-193 CD16 Mono MA0690.1 0.8896766 2.483767e-191 0.7723682
2 CD16 Mono EOMES 0.5862998 3.802279e-99 CD16 Mono MA0800.1 0.8922904 7.043330e-194 0.7392951
3 CD16 Mono RUNX3 0.7182799 7.394573e-84 CD16 Mono MA0684.2 0.6753424 3.175696e-40 0.6968111
motif.name <- ConvertMotifID(pbmc, name = 'SPI1')
gene_plot <- FeaturePlot(pbmc, features = "sct_SPI1", reduction = 'wnn.umap')
motif_plot <- FeaturePlot(pbmc, features = motif.name, min.cutoff = 0, cols = c("lightgrey", "darkred"), reduction = 'wnn.umap')
gene_plot | motif_plot
> # identify top markers in other cell types
> head(topTFs("Naive B"), 3)
RNA.group gene RNA.auc RNA.pval motif.group motif.feature motif.auc motif.pval avg_auc
1 Naive B EBF1 0.9604105 0.000000e+00 Naive B MA0154.4 0.7562941 8.038044e-26 0.8583523
2 Naive B POU2F2 0.6338272 2.833732e-09 Naive B MA0507.1 0.9702895 7.975628e-83 0.8020583
3 Naive B TCF4 0.7139073 2.689186e-41 Naive B MA0830.2 0.8641510 2.153784e-50 0.7890291
> head(topTFs("HSPC"), 3)
RNA.group gene RNA.auc RNA.pval motif.group motif.feature motif.auc motif.pval avg_auc
1 HSPC SOX4 0.9869221 7.766427e-71 HSPC MA0867.2 0.6967552 5.188151e-04 0.8418387
2 HSPC GATA2 0.7884615 0.000000e+00 HSPC MA0036.3 0.8240827 1.084319e-08 0.8062721
3 HSPC MEIS1 0.8830582 0.000000e+00 HSPC MA0498.2 0.7048912 3.010760e-04 0.7939747
> head(topTFs("Plasma"), 3)
RNA.group gene RNA.auc RNA.pval motif.group motif.feature motif.auc motif.pval avg_auc
1 Plasma IRF4 0.8189901 5.206829e-35 Plasma MA1419.1 0.9821051 1.452382e-12 0.9005476
2 Plasma MEF2C 0.9070644 4.738760e-12 Plasma MA0497.1 0.7525389 2.088028e-04 0.8298016
3 Plasma TCF4 0.8052937 5.956053e-12 Plasma MA0830.2 0.7694984 7.585013e-05 0.7873960
https://satijalab.org/seurat/v4.0/weighted_nearest_neighbor_analysis.html
https://monkeylearn.com/blog/what-is-tf-idf/
https://www.biorxiv.org/content/10.1101/460147v1.full
http://www.ludowaltman.nl/slm/#:~:text=The%20SLM%20algorithm%20has%20been%20implemented%20in%20the,be%20run%20on%20any%20system%20with%20Java%20support.
https://www.cwts.nl/blog?article=n-r2u2a4
https://github.com/chen198328/slm