TITLE: 全外显子测序数据分析
AUTHOR: Shuntai Yu
[TOC]
外显子是形成mRNA后剪接剩下的部分,包括UTR区与CDS,而CDS则是真正编码蛋白的Coding Sequence;

P1 —P4
这一系列视频比较基础,下游分析只讲了比较简单的注释,注释之后也只是看一下在外显子的突变。gatk的CNV变异等等都没有讲。
配套的参考代码参考文章1[1]和文章2[2],我觉得文章2更详细。
外显子只是DNA测序中的一个特例,是一种特殊的捕获测序,捕获芯片牌子很多,有安捷伦、illumina、罗氏。全基因组测序也是DNA测序,学会WES测序其它DNA测序也是差不多的。
文献1[3]一步一步讲的很细,从数据怎样出来一直到用到了哪些软件都讲了。随便找的文献2[4]和文献3[5],文献3啥也没发现,因为外显子是有将近一半的概率是找不到致病突变的,所以不好找。
收集2010至2018年的WES技术综述, 阅读超过5个公司的WES数据分析结题报告 :比如报告1[6]和报告2[7]。CCDS数据库就是人类所有外显子所在的地方,通过综述和结题报告形成对外显子测序的初步了解。
两个遗传疾病家系WES数据分析实战(曾提供给学员的数据)
首先要了解临床应用,可以仔细阅读 基因大讲堂全年知识点精华版: https://mp.weixin.qq.com/s/kysfshK55DA31xomucgRUA
准备:外显子相关软件和资料下载
软件下载
有些软件需要使用conda装,有些软件需要手动下载。
1.conda安装
参考文章1[1]
2.手动下载
gatk没有使用conda下载,因为曾想演示gatk4的用法,而conda没有gatk4,所以他使用谷歌搜索的gatk官网,使用wget进行的下载。下载过程有了一个解说很有意思

曾说他们办了100M的电信宽带所以下载快,图中阴影部分表示从亚马逊云下载的,从亚马逊云下载比较慢,但是可能是用的是亚马逊云的中国节点,所以下的比较快。
谷歌搜索gatk,曾使用的FilleZilla将下载到windows上的gatk-4.0.6.0.zip上传到了服务器,曾说因为在同一个路由下面所以上传的速度非常快。
当前做人相关的基因组分析,包括全基因组WGS,全外显子WES以及目标区域测序TRS,基本上都采用GATK标准的Best Practise最佳实践指导。人的基因组分析与其他物种稍微有一些不同,处理下载参考序列,还需要下载已有信息,例如各种人类基因组计划累积的变异信息,这些信息可以用于先验的学习集,提高变异检测的准确性。因此,如果想使用GATK软件,首先就需要下载GATK使用的数据集。所以gatk本身是完全不够的,还要下载100G的必备数据才能使用GATK,GATK在官网提供了一个resource bundle,里面包含了所需要的很多数据,如果使用gatk软件,最好把这些数据下载下来。
由于人的染色体存在多个数据中心,并且有多次更新,因此,当前有多个命名以及多个版本,搞不清这些版本和命名,有时候会产生很严重的后果。因为不同的命名之间序列的ID不同,比如1号染色体,有些是chr1,有些直接就是数字1,不同版本之间存在坐标这件的不同,因为很多注释数据库对染色体有版本要求,不同版本之间需要坐标转换之后才能使用。
这三种命名方式有一个对应关系。如下图所示.
| UCSC命名 | NCBI命名 | ENSEMBL命名 |
|---|---|---|
| hg18 | GRCh36 | ENSEMBL release_52 |
| hg19 | GRCh37 | ENSEMBL release_59/61/64/68/69/75 |
| hg38 | GRCh38 | ENSEMBL release_76/77/78/80/81/82 |
使用最多的就是hg19和hg38版本,理论上基因组版本越大,序列准确性越高。但是这里面有一个坐标位置的问题。就是我们无法保证坐标完全一致,基因组上坐标修改一个位置,与之相关联的所有内容要发生变化,例如这个坐标已经与dbSNP的rs号相对应了,这也就是为什么虽然现在已经有了hg38,但是hg19这个版本使用依然非常广泛,就是因为大量的注释信息都是基于hg19的版本来做的,如果要切换到hg38,所有的内容都需要改,工作量很大。此外,同一个版本还有很多子版本,例如37.1,37.2,37.3等。这种版本主要是基因组注释信息在更新,基因组序列没有发生变化。
GATK在官网提供了一个resource bundle,里面包含了所需要的很多数据,如果使用gatk软件,要把这些数据下载下来。有 FTP 和 Google Cloud bucket两种下载方式。但是google一般用不了,所以只能使用ftp的方式下载。
以下为100G的必备数据需要下载,下载的大小一定要对应。每个物种30多个G所以如果只做一个物种下30多G就好了
/public/biosoft/GATK/resources/bundle/hg38/bwa_index/
|-- [ 20K] gatk_hg38.amb
|-- [445K] gatk_hg38.ann
|-- [3.0G] gatk_hg38.bwt
|-- [767M] gatk_hg38.pac
|-- [1.5G] gatk_hg38.sa
|-- [6.2K] hg38.bwa_index.log
`-- [ 566] run.sh
/public/biosoft/GATK/resources/bundle/hg38/
|-- [1.8G] 1000G_phase1.snps.high_confidence.hg38.vcf.gz
|-- [2.0M] 1000G_phase1.snps.high_confidence.hg38.vcf.gz.tbi
|-- [3.2G] dbsnp_146.hg38.vcf.gz
|-- [3.0M] dbsnp_146.hg38.vcf.gz.tbi #我下的只有2.4M???重新下!
|-- [ 59M] hapmap_3.3.hg38.vcf.gz
|-- [1.5M] hapmap_3.3.hg38.vcf.gz.tbi
|-- [568K] Homo_sapiens_assembly38.dict
|-- [3.0G] Homo_sapiens_assembly38.fasta
|-- [157K] Homo_sapiens_assembly38.fasta.fai
|-- [ 20M] Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
|-- [1.4M] Mills_and_1000G_gold_standard.indels.hg38.vcf.gz.tbi
以上需要下载的东西也可以参考文章2[2]目录中所提到的文章2 《下载GATK需要的参考基因组文件》。
资料下载
主要是下载GATK需要的参考基因组文件,可以通过FTP下载,如下图所示

/resources/bundle/hg38/bwa_index/需要自己构建
软件安装之后是比对及IGV可视化,之后就是找变异。
第一步:QC
fastqc的命令很简单,直接跟文件即可,参数里面主要用-o(输出路径)和-t(线程,一般用2或4)
情况1:单个文件fastqc
QC涉及到的软件就是fastqc、trim_galore以及multiqc。 fastqc和multiqc的报告常常要整理成表格。
文件批量fastqc和multiqc
$ ls *.gz|xargs nohup fastqc -t 10 -o ../2020-4-16/qc/ & #xargs可以将前面的文件作为参数传给后面的命令,也可以用while read id do,不可以用管道,因为管道传的是文件名不是文件。
$ multiqc ./ #fastqc生成质控报告,multiqc将各个样本的质控报告整合为一个。 python3下无法运行multiqc,所以需要先创建python2的wes环境
情况2:批量并行对多个文件fastqc
方式一
$ ls SRR85182*.gz | while read id;do (nohup fastqc $id &);done #do后面要运行的内容使用()括起来,表示执行()内的命令。
方式二
$ find SRR851819*.gz|xargs fastqc -t 20 #-t 20 一次运行20个文件。(-t 是线程) 根据服务器情况选择,选择20个。
fastqc质控报告如何看自己去查!
第二步: trim
trim就是过滤低质量reads和去除接头,当质量差的时候这一步是必要的。使用的软件是trim-galore。软件的名字叫做trim-galore, 但是安装成功之后可执行文件是trim_galore二者中间的横不一样。如果没有接头就不用trim了,trim非常耗时。如果有adapter,trim之后再打开看会发现reads会变短,因为去掉了接头。
# 运行如下代码,得到名为config的文件,包含两列数据(双端测序需要的)
$ ls /data1/lixianlong/wangxiao/WGS/*_R1.fastq.gz > 1
$ ls /data1/lixianlong/wangxiao/WGS/*_R2.fastq.gz > 2
$ paste 1 2 > config
### 打开文件 qc.sh ,并且写入内容如下:
$ cat > qc.sh
source activate wes
bin_trim_galore=trim_galore
dir=$wkd/clean #注意修改$wkd
cat config |while read id
do
arr=(${id})
fq1=${arr[0]}
fq2=${arr[1]}
echo $dir $fq1 $fq2 #这里echo一下是为了调试,如果这三个变量无法打出,则说明代码有问题。曾就在定义$dir的时候使用了dir= $wkd/clean,发现老是报错,echo一下发现没有$dir,因为定义它的时候=和$之间多了一个空格,所以这一步echo看似多余,其实有用。
nohup $bin_trim_galore -q 25 --phred33 --length 36 -e 0.1 --stringency 3 --paired -o $dir $fq1 $fq2 &
done
source deactivate
$ bash qc.sh
可以将得到的文件再进行fastqc和multiqc,和trim-galore之前的fastqc以及multiqc结果进行对比就可以看出差别,如果觉得不太满意可以再去调节参数。trim-galore之后得到的是干净的测序数据,就可以用来跑后面的流程了。
–quality:设定Phred quality score阈值,默认为20。
–phred33:选择-phred33或者-phred64,表示测序平台使用的Phred quality score。
–length:设定输出reads长度阈值,小于设定值会被抛弃。因为前面发现算上adapter的长度是63,所以这里 的length将阈值设为36,表示去掉两端adapter之后,长度小于36的会被丢弃。而如果算上adapter的长度是 150,一般会将这里的length设为50,意思是将两端adapter去掉之后,长度小于50的会被丢弃。
-e:不是特别所谓
–stringency:设定可以忍受的前后adapter重叠的碱基数,默认为1(非常苛刻)。可以适度放宽,因为后一 个adapter几乎不可能被测序仪读到。 曾解释为adapter比对到多少算adapter,这里可以写成3,4,5都可 以,后期可以慢慢调。
–adapter:输入adapter序列。也可以不输入,Trim Galore! 会自动寻找可能性最高的平台对应的adapter。 自动搜选的平台三个,也直接显式输入这三种平台,即–illumina、–nextera和–small_rna。
–paired:对于双端测序结果,一对reads中,如果有一个被剔除,那么另一个会被同样抛弃,而不管是否达 到标准。
–retain_unpaired:对于双端测序结果,一对reads中,如果一个read达到标准,但是对应的另一个要被抛 弃,达到标准的read会被单独保存为一个文件。
–gzip和–dont_gzip:清洗后的数据zip打包或者不打包。
–output_dir:输入目录。需要提前建立目录,否则运行会报错。
– trim-n : 移除read一端的reads
第三步:mapping
mapping就是比对,将测序数据比对到参考基因组。
| 比对目的 | 比对用到的软件 | 比对的策略 |
|---|---|---|
| • 将打断测序的reads比回参考基因组 | • bwa(Burrows-Wheeler Aligner对准器 ) | • index先建立索引 |
| • 得到比对结果sam文本,用于后续分析 | bwa 是一款将序列比对到参考基因组上的软件 | • mem算法(maximal exact matches) |
(一)对参考基因组建立索引
bwa软件的作用是将序列比对到参考基因组上,在比对之前,首先需要对参考基因组建立索引。
#1.建立索引
$ ln -s /data1/lixianlong/YST/gatk4/resources/bundle/hg38/Homo_sapiens_assembly38.fasta /data1/lixianlong/YST/gatk4/resources/bundle/hg38/hg38.fa #使用软链接时被链接文件要使用绝对路径
$ bwa index /data1/lixianlong/YST/gatk4/resources/bundle/hg38/hg38.fa/Homo_sapiens_assembly38.fasta
## 人类参考基因组 hg38.fa
使用Homo_sapiens_assembly38.fasta来建立索引,然后会生成5个文件,后缀分别为 .bwt / .pac / .ann / .amb / .sa
(二)进行比对
这个步骤很慢,即使只单独取出10000条reads,比对依然很慢!
对质量较好的测序数据进行比对,bwa的命令一定不要使用nohup。nohup 的输出信息会被bwa输出到目标文件,会影响后续步骤。由于比对比较耗时,为节省时间,可以提取出前一万行小文件进行比对。
比对的格式由四部分组成,如下所示
bwa mem **index ** **R1文件 ** **R2文件 **
注意! index文件虽然有有.bwt / .pac / .ann / .amb / .sa五个,但是只写.前面的部分。

提取小文件进行后续操作(实战中可略步骤!)
情况1. 单个进行比对
$ source activate wes
$ find /data1/lixianlong/wangxiao/WGS -name *.gz >fq.txt #这一步成功执行的前提是不能当前路径不能是/data1/lixianlong/wangxiao/WGS
$ cat fq.txt |while read id; do(zcat $id|head -10000 > $(basename $id ".gz")); done # 得到的是小文件,然后使用这些小文件去进行mapping。$()表示运行()里面的代码
$ bwa #查看bwa手册
$ bwa mem #根据提示,当使用mem算法时候的手册
$ #bwa mem /data1/lixianlong/YST/gatk4/resources/bundle/hg38/hg38.fa/gatk_hg38 WGS-PDAC-sun_R1.fastq WGS-PDAC-sun_R2.fastq > WGS-PDAC-sun.sam #这个过程需要先将3G的索引加载到内存里面去,所以慢。
$ #bwa mem /data1/lixianlong/YST/gatk4/resources/bundle/hg38/hg38.fa/gatk_hg38 WGS-PDAC-sun_R1.fastq WGS-PDAC-sun_R2.fastq | samtools sort -@ 5 -o WGS-PDAC-sun.bam - #可以定位到文件的除了>还有管道,-@表示线程数,最后的-表示占位,因为让管道输出的是内容,内容需要占位。(发现不加-也行)
$ INDEX='/data1/lixianlong/YST/gatk4/resources/bundle/hg38/hg38.fa/gatk_hg38'
$ sample='WGS-PDAC-sun'
$ bwa mem -t 5 -R "@RG\tID:$sample\tSM:$sample\tLB:WGS\tPL:Illumina" $INDEX WGS-PDAC-sun_R1_val_1.fq.gz WGS-PDAC-sun_R2_val_2.fq.gz| samtools sort -@ 5 -o WGS-PDAC-sun.bam -
# -t和-R是bwa mem的参数,-t表示线程; 要求生成的文件是bam形式,所以不能使用>,而是使用samtools sort -o,这样生成的文件还是bam。
$ sample='WGS-hPC-P3'
$ bwa mem -t 5 -R "@RG\tID:$sample\tSM:$sample\tLB:WGS\tPL:Illumina" $INDEX WGS-hPC-P3_R1_val_1.fq.gz WGS-hPC-P3_R2_val_2.fq.gz| samtools sort -@ 5 -o WGS-hPC-P3.bam -
$ sample='WGS-hPC-P6'
$ bwa mem -t 5 -R "@RG\tID:$sample\tSM:$sample\tLB:WGS\tPL:Illumina" $INDEX WGS-hPC-P6_R1_val_1.fq.gz WGS-hPC-P6_R2_val_2.fq.gz| samtools sort -@ 5 -o WGS-hPC-P6.bam -
$ sample='WGS-hPC-P9'
$ bwa mem -t 5 -R "@RG\tID:$sample\tSM:$sample\tLB:WGS\tPL:Illumina" $INDEX WGS-hPC-P9_R1_val_1.fq.gz WGS-hPC-P9_R2_val_2.fq.gz| samtools sort -@ 5 -o WGS-hPC-P9.bam -
情况2. 批量比对
#首先构建一个文件,我们取名config
$ ls *R1_val_1.fq.gz >1
$ ls *R2_val_2.fq.gz >2
$ paste 1 2 > tmp
$ cat tmp |cut -f1|cut -d"_" -f1 > del
$ paste del tmp > config
$ cat config
WGS-PDAC-sun WGS-PDAC-sun_R1_val_1.fq.gz WGS-PDAC-sun_R2_val_2.fq.gz
WGS-hPC-P3 WGS-hPC-P3_R1_val_1.fq.gz WGS-hPC-P3_R2_val_2.fq.gz
WGS-hPC-P6 WGS-hPC-P6_R1_val_1.fq.gz WGS-hPC-P6_R2_val_2.fq.gz
WGS-hPC-P9 WGS-hPC-P9_R1_val_1.fq.gz WGS-hPC-P9_R2_val_2.fq.gz
$ INDEX='/data1/lixianlong/YST/gatk4/resources/bundle/hg38/hg38.fa/gatk_hg38'
$ cat config |while read id
$ do
#对config中的每一列进行赋值,并且开始循环读取和批量转换。
$ arr=($id)
$ fq1=${arr[1]} #fq1为第2列
$ fq2=${arr[2]} #fq2为第3列
$ sample=${arr[0]} #sample为第1列
$ bwa mem -t 5 -R "@RG\tID:$sample\tSM:$sample\tLB:WGS\tPL:Illumina" $INDEX $fq1 $fq2 | samtools sort -@ 5 -o $sample.bam -
$ done
讨论和思考?
为何要加 -R "@RG\tID:sample\tLB:WGS\tPL:Illumina" ? 这个问题很重要,我当时就犯了错,通过samtools view -H WGS-PDAC-sun.bam|grep -v "@SQ"才发现加和不加-R的差别。两种情况的对比如下:
若不加-R选项:
$ bwa mem $INDEX WGS-PDAC-sun_R1.fastq WGS-PDAC-sun_R2.fastq | samtools sort -@ 5 -o WGS-PDAC-sun_2.bam -
#查看bam文件;加了-R后这里会多一行@RG,若这行信息缺失则gatk会报错。但不加并不影响Samtools和VCF的使用。
$ samtools view -H WGS-PDAC-sun.bam|grep -v "@SQ"
@HD VN:1.6 SO:coordinate
@RG ID:WGS-PDAC-sun SM:WGS-PDAC-sun LB:WGS PL:Illumina
@PG ID:bwa PN:bwa VN:0.7.17-r1188 CL:bwa mem -R @RG\tID:\tSM:\tLB:WGS\tPL:Illumina /data1/lixianlong/YST/gatk4/resources/bundle/hg38/hg38.fa/gatk_hg38 WGS-PDAC-sun_R1.fastq WGS-PDAC-sun_R2.fastq
若加-R选项:
$ bwa mem -R "@RG\tID:$sample\tSM:$sample\tLB:WGS\tPL:Illumina" $INDEX WGS-PDAC-sun_R1.fastq WGS-PDAC-sun_R2.fastq | samtools sort -@ 5 -o WGS-PDAC-sun.bam -
#查看bam文件;不加-R后这里会少一行@RG
$ samtools view -H WGS-PDAC-sun_2.bam|grep -v "@SQ"
@HD VN:1.6 SO:coordinate
@PG ID:bwa PN:bwa VN:0.7.17-r1188 CL:bwa mem /data1/lixianlong/YST/gatk4/resources/bundle/hg38/hg38.fa/gatk_hg38 WGS-PDAC-sun_R1.fastq WGS-PDAC-sun_R2.fastq
比对的正常流程
不推荐单个比对,太慢!所以采用批量比对,如下所示:
#首先构建一个文件,我们取名config
$ ls *R1_val_1.fq.gz >1
$ ls *R2_val_2.fq.gz >2
$ paste 1 2 > tmp
$ cat tmp |cut -f1|cut -d"_" -f1 > del
$ paste del tmp > config
$ cat config
WGS-PDAC-sun WGS-PDAC-sun_R1_val_1.fq.gz WGS-PDAC-sun_R2_val_2.fq.gz
WGS-hPC-P3 WGS-hPC-P3_R1_val_1.fq.gz WGS-hPC-P3_R2_val_2.fq.gz
WGS-hPC-P6 WGS-hPC-P6_R1_val_1.fq.gz WGS-hPC-P6_R2_val_2.fq.gz
WGS-hPC-P9 WGS-hPC-P9_R1_val_1.fq.gz WGS-hPC-P9_R2_val_2.fq.gz
$ INDEX='/data1/lixianlong/YST/gatk4/resources/bundle/hg38/hg38.fa/gatk_hg38'
$ cat config |while read id
$ do
#对config中的每一列进行赋值,并且开始循环读取和批量转换。
$ arr=($id)
$ fq1=${arr[1]} #fq1为第2列
$ fq2=${arr[2]} #fq2为第3列
$ sample=${arr[0]} #sample为第1列
$ bwa mem -t 5 -R "@RG\tID:$sample\tSM:$sample\tLB:WGS\tPL:Illumina" $INDEX $fq1 $fq2 | samtools sort -@ 5 -o $sample.bam -
$ done
第四步:找变异
所有样本找变异
方法一:一步法找基因变异流程(未去除PCR重复)
这种方法比较简单,使用**samtools + bcftools **就可以
(一)查看bam文件
$ samtools mpileup WGS-PDAC-sun.bam
该命令就是针对bam文件的。打开之后具体每一列代表什么意思自己去查!这里尽量不要使用WGS-PDAC-sun.bam,因为它是抽出来的10000条reads转化的bam文件,没有代表性,可以使用真正的bam; samtools mpileup和samtools tview一样的效果,得到每个坐标的比对情况。去查看samtools mpileup的说明书
(二)bam文件转成VCF文件
$ ref=/data1/lixianlong/YST/gatk4/resources/bundle/hg38/Homo_sapiens_assembly38.fasta
$ source activate wes
$ time samtools mpileup -ugf $ref *.bam | bcftools call -vmO z -o out.vcf.gz
产生的VCF文件可以载入IGV里查看,载入之前需要先构建索引。 我没有成功运行,为啥呢??? 使用完整的bam好像又运行成功了!这一步也挺耗时!上面那一步首先将bam文件转成mpileup格式,然后bcftools用了call命令,call命令又有-vmO这些参数,最后生成了out.vcf.gz文件
mpileup参数的意思
-u 表示不要压缩,因为我们要通过管道直接把数据给bcftools的。
-g 输出到bcf格式给bcftools软件来做input。
-f 来输入有索引文件的fasta参考序列。
mpileup以前为pileup,如果大家看到的教程比较陈旧,会有这样的疑问。
samtools mpileup和bcftools call的具体参数意义见文章3[8]:
(三)查看VCF文件
$ less -S out.vcf.gz
产生的vcf文件会有很多头文件,因为参考基因组染色体特别多,具体vcf怎样看自己去查。产生的vcf文件可以载入到IGV里面,载入之前要先构建索引。
(四)批量构建索引
批量构建索引,为载入IGV做准备。这个过程比较耗时,产生的结果后缀为.bai
$ ls *.bam |xargs -i samtools index {}
这个流程没有去除PCR重复,只是简单的看一下,而去除PCR重复是非常必要的。但是对于转录组而言PCR重复是非常多的,但是转录组通常不会去除PCR重复。
(五)载入IGV查看
首先要把VCF文件下载到windows电脑上
(附1):去除PCR重复方法(samtools markdup -r)
去除PCR重复前和去除PCR重复后都有一个bam文件,去除PCR重复很耗时。去除PCR重复需要samtools的4个命令。
$ samtools #查找samtools用法
$ samtools markdup #查找samtools markdup用法,发现-r是去除PCR重复
$ samtools markdup -r WGS-PDAC-sun.bam WGS-PDAC-sun.rm.bam #该句运行失败! 但是曾这一步并没有报错。
上句代码运行报错如下[markdup] error: no ms score tag. Please run samtools fixmate on file first.,
解决方案参考文章2[2]里面的目录文章《5 一步法找基因变异流程》。
samtools markdup的语法格式是
samtools markdup
即需要一个input 和 output。其中option的-r、表示如果有duplicate就去除。 -S表示mark一下,这样后续的软件就知道它是duplicate
1. samtools sort
The first sort can be omitted if the file is already name ordered
$ cat sort.sh
samtools sort -n -o WGS-PDAC-sun.sort.bam WGS-PDAC-sun.bam
$ qsub -cwd -l vf=8g,p=8 -V sort.sh
2. samtools fixmate
Add ms and MC tags for markdup to use later
$ cat > fixmate.sh
samtools fixmate -m WGS-PDAC-sun.sort.bam WGS-PDAC-sun.fixmate.bam
$ qsub -cwd -l vf=8g,p=8 -V fixmate.sh
3. samtools sort
Markdup needs position order
$ cat > sort_2.sh
samtools sort -o WGS-PDAC-sun.positionsort.fixmat.bam WGS-PDAC-sun.fixmate.bam #samtools sort默认的通常是参考基因组坐标来进行的
$ qsub -cwd -l vf=8g,p=8 -V sort_2.sh
4. samtools markdup
Finally mark duplicates
$ cat > markdup_r.sh
samtools markdup -r WGS-PDAC-sun.positionsort.fixmat.bam WGS-PDAC-sun.rm.bam
$ qsub -cwd -l vf=8g,p=8 -V markdup_r.sh
对每一个bam文件都跑一下去除PCR重复的过程,得到rm.bam。
5. 比较rm前后的异同
以WGS-PDAC-sun.bam和WGS-PDAC-sun.rm.bam二者为例,查看rm前后是否有区别
$ samtools view WGS-PDAC-sun.rm.bam |less -S #查看下rm.bam
$ samtools flagstat WGS-PDAC-sun.bam #查看 WGS-PDAC-sun.bam 的reads数,若bam文件比较大,打开比较耗时,别急。
# 21430752 + 0 in total (QC-passed reads + QC-failed reads) #表示21430752条reads
# ... #其它的表示什么意思自己去查
$ samtools flagstat WGS-PDAC-sun.rm.bam #查看去除duplicate之后的reads数(duplicate就是指PCR重复)
# 18705712 + 0 in total (QC-passed reads + QC-failed reads) #表示去除PCR重复之后有18705712条reads
# ...
(附2):标记PCR重复方法(samtools markdup -S)
对PCR重复的reads进行标记的方法使用samtools markdup -S
$ samtools markdup -S WGS-PDAC-sun.positionsort.fixmat.bam WGS-PDAC-sun.mk.bam
$ samtools markdup -S WGS-hPC-P3.positionsort.fixmat.bam WGS-hPC-P3.mk.bam
$ samtools markdup -S WGS-hPC-P6.positionsort.fixmat.bam WGS-hPC-P6.mk.bam
$ samtools markdup -S WGS-hPC-P9.positionsort.fixmat.bam WGS-hPC-P9.mk.bam
查看WGS-PDAC-sun.bam和WGS-PDAC-sun.mk.bam二者区别
$ samtools view WGS-PDAC-sun.bam |less -S
$ samtools view WGS-PDAC-sun.mk.bam |less -S
查看是否加上@RG信息行
$ samtools view -H WGS-PDAC-sun.rm.bam|grep -v "@SQ" #查看是否加上@RG信息行
# @HD VN:1.6 SO:coordinate
# @RG ID:WGS-PDAC-sun SM:WGS-PDAC-sun LB:WGS PL:Illumina
# @PG ID:bwa PN:bwa VN:0.7.17-r1188 CL:bwa mem -t 5 -R @RG\tID:WGS-PDAC-sun\tSM:WGS-PDAC-sun\tLB:WGS\tPL:Illumina /data1/lixianlong/YST/gatk4/resources/bundle/hg38/hg38.fa/gatk_hg38 WGS-PDAC-sun_R1_val_1.fq WGS-PDAC-sun_R2_val_2.fq
$ samtools view -H WGS-hPC-P3.rm.bam|grep -v "@SQ"
$ samtools view -H WGS-hPC-P6.rm.bam|grep -v "@SQ"
$ samtools view -H WGS-hPC-P9.rm.bam|grep -v "@SQ"
方法二:GATK找变异
只有GATK才是真正的找变异
P9 wes-8-GATK4流程之标记PCR重复reads
P10 wes-9-GATK4流程之碱基质量值矫正上
P11 wes-10-GATK4流程之碱基质量值矫正下
曾主要参考的《GATK4操作文档》

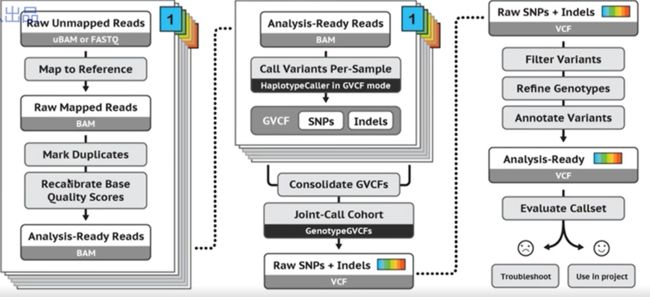
方法(一):使用for循环实现GATK
MarkDuplicates:标记PCR重复reads
FixMateInformation 和 index
BaseRecalibrator 和 ApplyBQSR:碱基矫正,BaseRecalibrator计算出了所有需要进行重校正的read和特征值,然后把这些信息输出为一份校准表文件(sample_name.recal_data.table)
BaseRecalibrator 和 ApplyBQSR包含了两个步骤:
第一步,BaseRecalibrator,这里计算出了所有需要进行重校正的read和特征值,然后把这些信息输出 为一份校准表文件(sample_name.recal_data.table)
第二步,PrintReads,这一步利用第一步得到的校准表文件(sample_name.recal_data.table)重新调 整原来BAM文件中的碱基质量值,并使用这个新的质量值重新输出一份新的BAM文件。
注意,因为BQSR实际上是为了(尽可能)校正测序过程中的系统性错误,因此,在执行的时候是按照不同的测序lane或者测序文库来进行的,这个时候@RG信息(BWA比对时所设置的)就显得很重要了,算法就是通过@RG中的ID来识别各个独立的测序过程,这也是我开始强调其重要性的原因。
HaplotypeCaller
可以将下面的代码包装进一个文件,命名为gatk.sh;但是有一点一定要注意,就是source activate wes这句代码的成功运行有几点是要注意的,如下所示:

#开始运行这一步,或者
source activate wes
ref=/data1/lixianlong/YST/gatk4/resources/bundle/hg38/Homo_sapiens_assembly38.fasta
snp=/data1/lixianlong/YST/gatk4/resources/bundle/hg38/dbsnp_146.hg38.vcf.gz
indel=/data1/lixianlong/YST/gatk4/resources/bundle/hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
for sample in {WGS-PDAC-sun,WGS-hPC-P3,WGS-hPC-P6,WGS-hPC-P9}
do
echo $sample
# Elapsed time: 7.91 minutes #对照着知道大概时间多久防止出错。
gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" MarkDuplicates \
-I $sample.bam \
-O ${sample}_marked.bam \
-M $sample.metrics \
1>${sample}_log.mark 2>&1
#这里产生的输出是FixMateInformation那一步的input;#曾说-M可以输也可以不输; # 1>表示输到桌面上的;2>表示错误输出; 使用2>&1是因为认为不会有2所以才将2输入到1里面去; #曾说java软件每次启动都需要创建一个虚拟机,叫做java虚拟机,java软件都是在java虚拟机上运行的。这样一个软件在什么系统都可以使用,windows,mac等等;
## Elapsed time: 13.61 minutes
gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" FixMateInformation \
-I ${sample}_marked.bam \
-O ${sample}_marked_fixed.bam \
-SO coordinate \
1>${sample}_log.fix 2>&1
#进行index
samtools index ${sample}_marked_fixed.bam
## 17.2 minutes #这一步我卡了好久,运行老是出错! #我使用conda安装gatk发现成功了。借鉴的《gatk BaseRecalibrator 报错》https://www.jianshu.com/p/ddab0008d792
# $ conda install gatk4=4.0.6.0
gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" BaseRecalibrator \
-R $ref \
-I ${sample}_marked_fixed.bam \
--known-sites $snp \
--known-sites $indel \
-O ${sample}_recal.table \
1>${sample}_log.recal 2>&1
gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" ApplyBQSR \
-R $ref \
-I ${sample}_marked_fixed.bam \
-bqsr ${sample}_recal.table \
-O ${sample}_bqsr.bam \
1>${sample}_log.ApplyBQSR 2>&1
## 使用GATK的HaplotypeCaller命令
gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" HaplotypeCaller \
-R $ref \
-I ${sample}_bqsr.bam \
--dbsnp $snp \
-O ${sample}_raw.vcf \
1>${sample}_log.HC 2>&1
done
$ conda deactivate #先失活wes这一conda环境
$ qsub -cwd -l vf=8g,p=8 -V gatk.sh
方法(二):使用while循环实现GATK
$ cat > config
WGS-PDAC-sun
WGS-hPC-P3
WGS-hPC-P6
WGS-hPC-P9
$ cat config|while read id
$ do
$ sample=id #下面的sample就不用全部改成id了
$ echo $sample
... #嵌入的内容
$ done
$ GATK=/data1/lixianlong/YST/gatk4/gatk-4.0.6.0/gatk
$ $GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" MarkDuplicates #注意是$GATK
$ 运行上面两行代码就可以看到说明书,其中Required Arguments:就是必须要选的,Optional Arguments:是非必须的。--REMOVE_SEQUENCING_DUPLICATES:Boolean如果选TRUE则会去掉一些duplicate,bam文件就会变小,如果没有选TRUE则只会对duplicate标记一下。使用相同的方法也可以另外几步说明书
$ GATK=/data1/lixianlong/YST/gatk4/gatk-4.0.6.0/gatk
$ sample=WGS-PDAC-sun
$ nohup $GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" MarkDuplicates -I $sample.bam -O ${sample}_marked.bam -M $sample.metrics 1>${sample}_log.mark 2>&1 &
$ top -u lixianlong #可看到有java程序在运行,因为gatk是一个java软件
$ ps -ef|grep "java" #从该命令的结果可见,虽然很多参数没有加但是却会默认加上。
以上就是GATK的上游流程,通过以上步骤得到所有样本的vcf文件
对比看一下曾的结果,看后删掉!!!

(附2):并行和批处理异同
批处理和并行是不一样的;while read id; do ();done结构就是批处理,一个样本处理完之后再去处理第二个。而代码尾部加上&提交后台则属于并行;nohup表示不要关机,&表示提交到后台。
markduplicate这一步是一定要做的,可以使用picard或者samtools或者bwa都可以
通过搜索picard sam flag 进入网页 Explain SAM Flags 可以在该网页查询flag数字表示的含义
曾说之所以使用GATK去找变异是因为做人类的比较仔细,其实samtools + bcftools就可以简单找变异。GATK需要经过5个步骤。
第五步:对比对和变异结果用IGV进行可视化
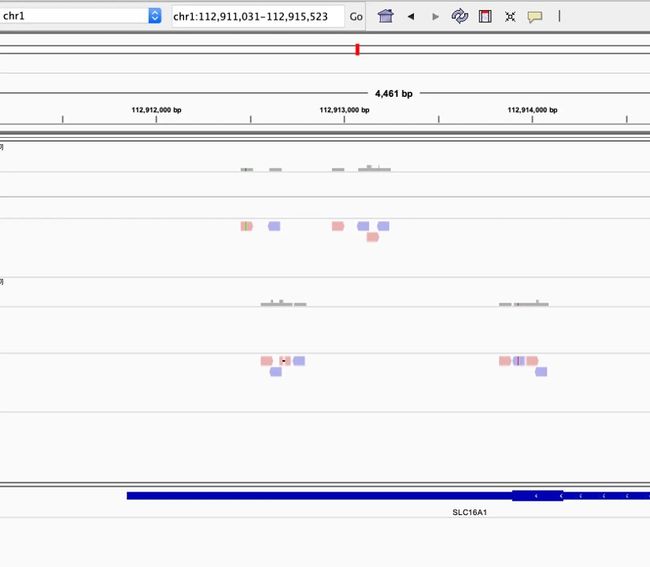
(一)检查感兴趣基因区域内比对和找变异情况(以BRCA1基因为例)
BaseRecalibrator得到bam之后,如果是tumor和normal则是两个bam文件,需要对两个bam文件进行处理进而得到vcf文件,这个vcf文件叫做somatic vcf。 germline vcf比较简单一些。
bqsr.bam要比其它bam大很多,因为BaseRecalibrator表示重新计算,本来就有一个质量值,通过重新计算再得到新的质量值,因此bqsr.bam比其它bam文件列数增多。事实上有参数可以只输出矫正后的值,然后弃去矫正前的值,这样文件就会小很多,这在处理大批量数据的时候很好用。
1. 下载gtf文件
gtf有不同版本???
$ wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_25/gencode.v25.annotation.gtf.gz
$ zcat gencode.v25.annotation.gtf.gz |grep BRCA1
$ wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_32/gencode.v32.annotation.gtf.gz
gunzip gencode.v32.annotation.gtf.gz
2. 找到BRCA1在gtf文件中的坐标
$ zcat gencode.v25.annotation.gtf.gz |grep -w BRCA1|head|less -SN #这步需要一定的时间
chr17 HAVANA gene 43044295 43170245 #得到BRCA1的坐标位于17号染色体,起始和终止坐标也知道了。
$ ls -lh WGS-PDAC-sun*bam
625K Apr 23 13:47 WGS-PDAC-sun.bam
1010K Apr 22 17:57 WGS-PDAC-sun_bqsr.bam
827K Apr 22 17:57 WGS-PDAC-sun_marked.bam
830K Apr 22 17:57 WGS-PDAC-sun_marked_fixed.bam
$ samtools index WGS-PDAC-sun.bam #先运行这一步,产生WGS-PDAC-sun.bai文件下一步才能运行成功
$ samtools view WGS-PDAC-sun.bam chr17:43044295-43170245 #但是即使上一步成功运行,这一步也可能没有任何反应,除非这里是用的WGS-PDAC-sun.bam是完整的,而不是抽出来的1000条reads。因为该区间对于小bam没找到。
$ samtools view WGS-PDAC-sun.bam chr17:43044295-43170245 |wc
911 15539 403770 #发现有911条reads
3. 根据基因坐标提取子集
根据基因坐标从bam文件提取子集,也就是提取出BRCA1基因的reads。 把WGS-PDAC-sun各个bam里的BRCA1基因的reads拿出来,如下所示:
$ samtools view -h WGS-PDAC-sun.bam chr17:43044295-43170245 |samtools sort -o WGS-PDAC-sun.brca1.bam -
$ samtools view -h WGS-PDAC-sun_bqsr.bam chr17:43044295-43170245 |samtools sort -o WGS-PDAC-sun_bqsr.brca1.bam -
$ samtools index WGS-PDAC-sun_marked.bam
$ samtools view -h WGS-PDAC-sun_marked.bam chr17:43044295-43170245 |samtools sort -o WGS-PDAC-sun_marked.brca1.bam -
$ samtools view -h WGS-PDAC-sun_marked_fixed.bam chr17:43044295-43170245 |samtools sort -o WGS-PDAC-sun_marked_fixed.brca1.bam -
samtools view是没有header的,想要看header要加-h,这样再samtools sort才能生成小bam。把这些bam里面的BRCA1基因的reads拿出来放在一个小bam文件中。要求拿出来之后还是bam形式,所以不能使用 >,而要是用samtools sort -o,这样生成的文件还是bam。曾说最新版的samtools sort就可以将sam转成bam,曾说排序就可以将sam转成bam了。
4. 构建index
为上一步骤产生的所有brca1.bam文件构建index
$ ls *brca1.bam|xargs -i samtools index {}
$ mv *brca1* tmp/
$ cd tmp
把tmp文件下载到windows 的IGV查看
注意,igv同时需要.bam和相应的.bai文件,所以需要把整个文件夹cp。
5. 使用IGV进行可视化
IGV启动每次都要加载一下基因组和注释文件,网速如果不好会很慢,可以将基因组和注释文件下载到电脑,然后从本地加载基因组。以人类基因组为例,操作如下:
# 最好先用samtools 的faidx构建一个.fai索引文件
# for hg19 genome
$ cd ~/reference/genome/hg19
$ wget -c http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/hg19.fa.gz
$ gunzip hg19.fa.gz && rm hg19.fa.gz
$ samtools faidx hg19.fa #构建索引,产生hg19.fa.fai
# for hg38 genome
$ cd ~/reference/genome/hg38
$ wget -c http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/hg38.fa.gz
$ gunzip hg38.fa.gz && rm hg38.fa.gz
$ samtools faidx hg38.fa #构建索引,产生hg38.fa.fai
把fa文件和它的索引.fai文件放在本地,然后只需要通过Genomes=>Load Genome from File,导入FASTA文件。

另外,还需要基因的注释,即加载基因组GTF文件
例如下载人类基因组基因注释文件:
$ wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_32/gencode.v32.annotation.gtf.gz
$ gunzip gencode.v32.annotation.gtf.gz
File => Load from file => 选择解压后的GTF文件,这是为了能看到基因的信息(如下图底部记录)
基因组和GTF文件都有了,就可以载入bam文件查看了,不过好像不能按照基因名查找,可以按照基因的坐标查找。
(二)针对特定基因找变异(以BRCA1基因为例)
$ source activate wes
$ ref=/data1/lixianlong/YST/gatk4/resources/bundle/hg38/Homo_sapiens_assembly38.fasta
方法一:一步法找变异
这种方法比较简单,使用**samtools + bcftools **就可以
有了这些特定基因区域的小bam文件,就可以利用这些小bam文件来找变异,因为这个小bam文件是只含有一个基因的小bam文件,所以找的变异是特定基因的变异。
$ samtools mpileup -ugf $ref WGS-PDAC-sun_bqsr.brca1.bam | bcftools call -vmO z -o WGS-PDAC-sun_bqsr.vcf.gz #曾说虽然warning,但不影响使用。
事实上不使用特定基因区域的小bam文件也可以针对特定基因找变异,只需要对samtools mpileup指定区域,然后就可以对大bam文件针对该区域来找变异。
所有样本走samtools mpileup 和bcftools call 流程
$ source activate wes
$ wkd=/data1/lixianlong/wangxiao/2020-4-16/small
$ cd $wkd/align
$ ls *_bqsr.bam |xargs -i samtools index {}
ref=/data1/lixianlong/YST/gatk4/resources/bundle/hg38/hg38.fa/Homo_sapiens_assembly38.fasta
$ nohup samtools mpileup -ugf $ref *_bqsr.bam | bcftools call -vmO z -o all_bqsr.vcf.gz &
方法二:GATK找变异
也可以使用前面GATK的五步法来得到vcf文件
代码参考前面
查看一下
$ zcat WGS-PDAC-sun_bqsr.vcf.gz
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT WGS-PDAC-sun #CHROM表示变异所在的染色体,POS表示变异的坐标(只能叫变异,不能叫突变,因为还要对vcf进行过滤、注释,这只能说明它和参考基因组不一样,而参考基因组本身有很多致病位点之类的。),其其它的自己查。
查看文章3[9]就可以明白这三个组学究竟是什么东西。
全基因组测所有地方;全外显子组只测外显子;转录组测外显子但是外显子和外显子是连接起来的,因为内含子被切掉了;chip-seq测跟蛋白结合的地方;
第六步:比对及找变异步骤的质控
raw和clean的fastq文件都需要使用fastqc质控。
比对的各个阶段的bam文件都可以质控。
使用qualimap对wes的比对bam文件总结测序深度及覆盖度。参考《qualimap+multiqc完美解决多组学比对结果的质控》
(一)对于比对各阶段产生的bam文件质控
1. 构建index
只留第一个(WGS-PDAC-sun.bam)和最后一个bam(WGS-PDAC-sun.bqsr.bam),中间过程没有什么意义,所以删掉。bam文件质控也需要建index,有了index之后才能对它进行各种各样的质控。
$ source activate wes
$ wkd=/data1/lixianlong/wangxiao/2020-4-16/small/small_fq
$ cd $wkd/clean
$ ls *.gz |xargs fastqc -t 10 -o ./
$ cd $wkd/align
$ rm *_marked*.bam
$ ls *.bam |xargs -i samtools index {} #构建index
2. 对bam文件质控
bam文件质控使用samtools flagstat
$ ls *.bam | while read id ;do (samtools flagstat $id > $(basename $id ".bam").stat);done # while read id; do (); done 结构。
$ cat WGS-PDAC-sun.stat #查看结果,可以将结果复制进表格,做成报表,类似于晶能生物结题报告3.2数据比对统计中1,比对统计文件。summary文件中原始碱基数单位G表示十亿,1G碱基表示十亿碱基。
# 4938 + 0 in total (QC-passed reads + QC-failed reads)
$ samtools view WGS-PDAC-sun.bam|wc -l
# 4938
(二)总结测序深度及覆盖度
到CCDS官网ftp.ncbi.nlm.nih.gov/pub/CCDS/下载CCDS序列数据。并自己制作mm10.exon.chr.bed(外显子坐标文件),做好之后就可以使用qualimap。CCDS是根据gtf来做的,每个文件要么可以自己下载(前提是你有足够的背景知识知道它存在),要么只要一个fastq文件,其它自己去做(前提是你编程很厉害),所有东西都可以自己做,但是参考基因组做不了,只能自己下载。如果将来去公司工作,一定要会脚本,因为公司的很多需求是现有的科研团体难以完成或不愿意去做的。
1. 下载CCDS序列并安装bedtools
$ wget ftp://ftp.ncbi.nlm.nih.gov/pub/CCDS//archive/20/CCDS.20160908.txt #下载CCDS.20160908.txt文件
$ conda install bedtools #因为已经设置好清华镜像了,所以安装比较快。conda镜像只需要设置一次???即使推出终端,下次再登依然有效???查一下!!!
$ wkd=/data1/lixianlong/wangxiao/2020-4-16/small/gatk
$ samtools view WGS-PDAC-sun.bam|less -S #我们可以看到染色体有chr,比如1号染色体是chr1。
$ cat CCDS.20160908.txt|less -S #可以看到染色体是是数字,比如1号染色体是1。

2.制作exon.bed文件
我们需要将CCDS.20160908.txt做成外显子坐标文件,也就是对CCDS.20160908.txt文件进行转换处理,比如将染色体从数字变成chr+数字,一行变成多行之类的。
$ cat CCDS.20160908.txt|perl -alne '{/\[(.*?)\]/;next unless $1;$gene=$F[2];$exons=$1;$exons=~s/\s//g;$exons=~s/-/\t/g;print "$F[0]\t$_\t$gene" foreach split/,/,$exons;}'|sort -u |bedtools sort -i |awk '{print "chr"$0"\t0\t+"}' > $wkd/hg38.exon.bed
解释一下这个命令吧:
首先把以#开头的注释信息去除 grep -v '^#'
然后用正则匹配抓取中括号里面的信息 /[(.*?)]/
接着对中括号里面的信息依据逗号进行分割成数组 split/,/,len+=(tmp[0])
然后判断该转录本的外显子长度之和是否大于记录到该转录本对应的基因记 录的长度,如果大于,就重新记录下来,因为我们要取最长转录本。 F[2]}=len >F[2]}
最后把记录好的基因对应的最长转录本的外显子长度之和打印出来即可 print "h{$_}" foreach sort keys %h}'
$ cat hg38.exon.bed|less -S #可以看到染色体从数字变成了chr+数字;一个基因出现了多次,是因为一行变成了多行,本来一行有好多外显子区间,转换之后每个外显子区间都单独放一行。并且还可以看到有6列,有一列全部是0,还有一列全部是+。这两列是没有任何卵用的,纯粹是为了凑数,因为bed文件必须是3列或者6列才可以,4列是不行的,所以弄了一列0和一列+来凑数。其实qualimap真正用到的只有三列。
$ exon_bed=hg38.exon.bed
$ mkdir stats; cd stats
$ mv ../*.stat ./
$ multiqc ./ #这一句我运行时出现无法运行的问题,主要是python的版本,我将conda的wes环境失活,使用conda的base环境,发现运行这句代码成功了。 将产生的文件下载到本地查看。
#很奇怪!为什么 M Reads Mapped都是0?????TMD
3. qualimap进行质控
$ ls *_bqsr.bam | while read id;
$ do
$ sample=${id%%.*} # .*就是把.后的东西去掉。
$ echo $sample
$ qualimap bamqc --java-mem-size=20G -gff $exon_bed -bam $id &
$ done
$ mkdir qualimap; cd qualimap
$ mv ../*bqsr_stats ./
$ multiqc ./ #可以对qualimap的结果也进行multiqc,查看结果,外显子测序要求coverage都大于30X才有意义。我发现我的结果还是有问题!!!
### featureCounts
gtf=/data1/lixianlong/wangxiao/2020-4-16/gtf/gencode.v25.annotation.gtf.gz
featureCounts -T 5 -p -t exon -g gene_id \
-a $gtf *_bqsr.bam -o all.id.txt 1>counts.id.log 2>&1 & #featureCounts可以使用的前提是安装了subread;featureCounts是根据bam文件和gtf文件来进行计数。
拿到vcf文件只是wes数据分析的开始,很多paper和公司会直接提供vcf文件,上游分析并不太重要,拿到vcf之后的东西才更重要,很多应用类的文章最后找不到致病位点;
(三)比较samtools和gatk找变异的区别
samtools找的变异
$ zcat out.vcf.gz |grep -w chr1|less
#底下这个是我从网上考的,并不是上面这句代码运行的结果。
# chr1 139213 . A G 388 . #这一部分描述的是哪个位置上的突变
# DP=984;VDB=0.831898;SGB=-223.781;RPB=0.599582;MQB=0.0001971;MQSB=0.00457372;BQB=2.59396e-09;MQ0F=0.281504;ICB=0.333333;HOB=0.5;AC=3;AN=6;DP4=371,169,160,84;MQ=21
# GT:PL 0/1:198,0,255 0/1:176,0,255 0/1:50,0,158 #这一部分是每个样本的信息。至于DP,VDB,SGB这些之类的都是啥意思可以从头文件查阅。
$ zcat out.vcf.gz |grep -v contig|less -S ##INFO=就是解释信息。
gatk找的变异
$ cat *.vcf|grep -w 139213 # 这个vcf是gatk得到的vcf
# chr1 139213 rs370723703 A G 3945.77 . AC=1;AF=0.500;AN=2;BaseQRankSum=-2.999;ClippingRankSum=0.000;DB;DP=244;ExcessHet=3.0103;FS=2.256;MLEAC=1;MLEAF=0.500;MQ=29.33;MQRankSum=-0.929;QD=16.17;ReadPosRankSum=1.462;SOR=0.863 GT:AD:DP:GQ:PL 0/1:136,108:244:99:3974,0,6459
# chr1 139213 rs370723703 A G 2261.77 . AC=1;AF=0.500;AN=2;BaseQRankSum=-1.191;ClippingRankSum=0.000;DB;DP=192;ExcessHet=3.0103;FS=9.094;MLEAC=1;MLEAF=0.500;MQ=32.03;MQRankSum=-0.533;QD=11.78;ReadPosRankSum=0.916;SOR=0.321 GT:AD:DP:GQ:PL 0/1:126,66:192:99:2290,0,7128
# chr1 139213 rs370723703 A G 2445.77 . AC=1;AF=0.500;AN=2;BaseQRankSum=-2.495;ClippingRankSum=0.000;DB;DP=223;ExcessHet=3.0103;FS=10.346;MLEAC=1;MLEAF=0.500;MQ=30.18;MQRankSum=0.486;QD=10.97;ReadPosRankSum=-0.808;SOR=0.300 GT:AD:DP:GQ:PL 0/1:152,71:223:99:2474,0,7901
曾说samtools找的变异是几个样本一起找,gatk找的变异是分开找。从位点的角度来说二者找的没有啥不一样。因为很多时候是不是变异通过IGV可视化都可以看出来,所以使用不同软件找变异按道理说不应该有所差异,但是有些变异通过肉眼判断就很难,比如10个reads有两个有变异,八个没变异,这个时候就不好判断有没有变异,因为逻辑思考没有一个很清晰的界限。虽然可以设定一个阈值为2,但是假如两个有变异的reads质量很差,则很可能是假阳性,所以要考虑的因素就会比较多,不同软件的标准很难全部一样,所以可能会有不同软件结果差异现象的存在。
《WES(五)不同软件比较》
补充作业
使用freebayes和varscan找变异。
VCF下游分析
主要是:注释和过滤
注释
VEP,snpEFF,ANNOVAR都可以注释
《【直播】我的基因组 32:使用annovar注释vcf》
《ANNOVAR 软件用法还可以更复杂》
(一)ANNOVAR介绍
1.Annovar使用记录 (http://www.bio-info-trainee.com/641.html)
2.用annovar对snp进行注释 (http://www.bio-info-trainee.com/441.html)
3.对感兴趣的基因call variation(http://www.bio-info-trainee.com/2013.html)
4.WES(六)用annovar注释(http://www.bio-info-trainee.com/1158.html)
ANNOVAR是一个perl程序,是高效的注释工具,能够利用最新的数据来分析各种基因组中的遗传变异。支持包括VCF在内的多种输入和输出文件格式。有三种注释方法:gene-based, region-based 和filter-based。
基于基因的注释(Gene-based Annotation)揭示variant与已知基因直接的关系以及对其产生的功能性影响;
基于区域的注释(Region-based Annotation)这个用到不多,揭示variant 与不同基因组特定段的关系,例如:它是否落在转录因子结合区域等。
基于数据库的过滤注释(Filter-based Annotation)这个很容易理解,则分析变异位点是否位于指定的数据库中,比如dbSNP, 1000G,ESP 6500等数据库,计算SIFT/PolyPhen/LRT/MutationTaster/MutationAssessor等。
(二)ANNOVAR软件下载安装
参考文章《ANNOVAR -注释软件用法详解》https://mp.weixin.qq.com/s?src=11×tamp=1588115119&ver=2307&signature=6NDdUhAZIRUTpbxLfshjPBC6j-LfL9XhyERlzgLfKnGx1DRO7IMRoEn9mWfCSdatYAy6wYhBOVF8RLL1EOOZMSfp1jPUxEVvJ7fNxPYoeVMgC-fUbf-Hhh0Neirn6CZr&new=1
官网下载中心:http://annovar.openbioinformatics.org/en/latest/user-guide/download/ 注册->网站发送邮件->直接邮件下载(需使用机构邮箱)->拷贝到服务器->tar解压安装
解压后生成annovar文件夹,里面有6个perl脚本程序和两个文件夹,其中一个是example文件夹,另一个是已经建立好的hg19或者GRCh37的humandb的数据库文件夹,可用于人的注释。
$ tar zxvf annovar.latest.tar.gz
解压后生成annovar文件夹,里面有6个perl脚本程序和两个文件夹,其中一个是example文件夹,另一个是已经建立好的hg19或者GRCh37的humandb的数据库文件夹,可用于人的注释。最重要的是humandb的文件夹,其中包括人类的数据库,是注释时必须的。
ANNOVAR结构
│ annotate_variation.pl #主程序,功能包括下载数据库,三种不同的注释
│ coding_change.pl #可用来推断突变蛋白质序列
│ convert2annovar.pl #将多种输入格式转为.avinput的程序
│ retrieve_seq_from_fasta.pl #用于自行建立其他物种的转录本
│ table_annovar.pl #一次可完成多种类型的注释
│ variants_reduction.pl #可用来更灵活地定制过滤注释流程
│
├─example #存放示例文件
│
└─humandb #人类注释数据库,全称human database,最重要!
(三)下载各种数据库(也就是各种资源库)
ANNOVAR可以下载很多数据库,通过-downdb参数直接从UCSC Genome Browser Annotation Database检索。此外,通过-downdb -webfrom annovar 参数可以从annovat提供的镜像下载人的数据库。我们可以自己下载所需数据库,数据库名称参考官网提供的数据库列表,套用代码。
1. 下载人类变异注释数据库
方法一:使用ANNOVAR下载
格式1: perl annotate_variation.pl --downdb 库名 --buildver 参考基因组版本 humandb/
格式2: perl annotate_variation.pl --downdb --webfrom annovar 库名 --buildver 参考基因组版本 humandb/ (格式2加上了镜像)。
#曾说因为是perl程序,所以要加perl,如果改了权限变成777就不需要加perl, 但是head annotate_variation.pl可以看到#!后有perl所以这里不加perl也是可以的。annotate_variation.pl后的.pl表示的就是perl程序的意思
-downdb表明该命令的用途是下载数据库;
#-buildver指定基因组版本(默认为hg18),参考基因组版本,通常选择hg38, 很少看到hg19了。
#-webform annovar 从annovar提供的镜像下载(不加此参数将默认从ucsc下载);
#refGene代表的是下载的数据库的名字;
humandb/表示数据库存储的路径;
# 进入软件安装目录运行(即annotate_variation.pl所在文件夹下运行)。
$ nohup ./annotate_variation.pl --downdb --webfrom annovar gnomad_genome --buildver hg38 humandb/ >down.log 2>&1 & #只要将该句代码gnomad_genome更改掉就可以了,更改成[官网提供的数据库列表]中所提供的其它数据库的名字即可!如下所示
$ nohup ./annotate_variation.pl --downdb --webfrom annovar refGene --buildver hg38 humandb/ >down.log 2>&1 &
# .
# .
# .
或者在任意地方运行如下代码:
$ perl /data1/lixianlong/wangxiao/2020-4-16/annovar/annotate_variation.pl -downdb -webfrom annovar gnomad_genome --buildver hg38 /data1/lixianlong/wangxiao/2020-4-16/annovar/humandb/
#只要将该句代码gnomad_genome更改掉就可以了,更改成[官网提供的数据库列表]中所提供的其它数据库的名字即可!如下所示
$ perl /data1/lixianlong/wangxiao/2020-4-16/annovar/annotate_variation.pl -downdb -webfrom annovar refGene --buildver hg38 /data1/lixianlong/wangxiao/2020-4-16/annovar/humandb/
# .
# .
# .
方法二:使用wget命令下载
$ wget -t 1 -T 30 -q -O hg38_gnomad_genome.txt.gz http://www.openbioinformatics.org/annovar/download/hg38_gnomad_genome.txt.gz
# hg38_gnomad_genome.txt大小是16G
# -t 设置重试次数,-T 设置超时为SECONDS秒,-q 静默下载(无信息输出),-O 将文档写入FILE
# 这个文件解压后有16G,注意云服务器的空间。
# 当然,注释上,并不等价于理解它们,这是很大的工作量。
通常要下载的数据库非常多,包括:dbSNP,ExAC,ESP6500,cosmic,gnomad,1000genomes,clinvar,gwas等。
# 'cytoBand' 每个细胞间 band(cytogenetic band)的染色体坐标信息
$ perl annotate_variation.pl --downdb --buildver hg19 cytoBand humandb/
# '1000g2015aug' alternative allele frequency data in 1000 Genomes Project(version August 2015)
$ perl annotate_variation.pl --downdb --webfrom annovar --buildver hg19 1000g2015aug humandb/
# 'exac03' ExAC 65000 exome allele frequency data for ALL, AFR (African), AMR (Admixed American), EAS (East Asian), FIN (Finnish), NFE (Non-finnish European), OTH (other), SAS (South Asian)). version 0.3. Left normalization done
$ perl annotate_variation.pl --downdb --webfrom annovar --buildver hg19 exac03 humandb/
# 'ljb26_all' whole-exome SIFT, PolyPhen2 HDIV, PolyPhen2 HVAR, LRT, MutationTaster, MutationAssessor, FATHMM, MetaSVM, MetaLR, VEST, CADD, GERP++, PhyloP and SiPhy scores from dbNSFP version 2.6
$ perl annotate_variation.pl --downdb --webfrom annovar --buildver hg19 ljb26_all humandb/
# 'clinvar_20200316' CLINVAR database with Variant Clinical Significance (unknown, untested, non-pathogenic, probable-non-pathogenic, probable-pathogenic, pathogenic, drug-response, histocompatibility, other) and Variant disease name
$ perl annotate_variation.pl --downdb --webfrom annovar --buildver hg19 clinvar_20200316 humandb/
# 'snp138' dbSNP with ANNOVAR index files
$ perl annotate_variation.pl --downdb --webfrom annovar --buildver hg19 snp138 humandb/
# databases not stored in UCSC or ANNOVAR-DB
$ perl annotate_variation.pl --downdb --webfrom http://www.regulomedb.org/downloads/ RegulomeDB.dbSNP141 /home/annovar/humandb
曾健明下载的东西很多,大小有41G,如下图所示:
(四)注释自己的vcf文件数据
vcf文件有三种注释方式,基于基因的注释、基于区域的注释、基于数据库的过滤
1. 基于基因的注释
基于基因的注释是想搞清楚自己的vcf文件记录的突变位点,是否在基因组的哪些功能元件(主要是外显子)上面
ANNOVAR基于基因的注释是下面三步
$ cd /data1/lixianlong/wangxiao/2020-4-16/biosoft
$ ln -s /data1/lixianlong/wangxiao/2020-4-16/annovar annovar #通过软连接做了一个非常巧妙的替换,使/data1/lixianlong/wangxiao/2020-4-16/biosoft路径下有该文件夹的链接,这样不用每次都改代码。
(1):转换vcf的格式
ANNOVAR主要使用convert2annovar.pl程序进行转换。
$ /data1/lixianlong/wangxiao/2020-4-16/biosoft/annovar/convert2annovar.pl -format vcf4old WGS-PDAC-sun_raw.vcf > WGS-PDAC-sun.annovar 2> /dev/null
$ /data1/lixianlong/wangxiao/2020-4-16/biosoft/annovar/convert2annovar.pl -format vcf4old WGS-hPC-P3_raw.vcf > WGS-hPC-P3.annovar 2> /dev/null
#我在vcf文件所在文件夹下执行的该命令。
# WGS-PDAC-sun.annovar就是从WGS-PDAC-sun_raw.vcf中提取信息
$ head WGS-PDAC-sun.annovar #可以看到把染色体、坐标、突变信息等信息提取了出来
(2):注释
注释通常是比较快的,因为perl程序通常都不会太慢。
$ /data1/lixianlong/wangxiao/2020-4-16/biosoft/annovar/annotate_variation.pl -buildver hg38 \
--geneanno \
--outfile WGS-PDAC-sun.anno \
WGS-PDAC-sun.annovar \
/data1/lixianlong/wangxiao/2020-4-16/biosoft/annovar/humandb/ #这一句是database,不可以少,曾把这里错当成另外一个命令给删掉了,他调了很久。
#上面的代码把WGS-PDAC-sun.annovar和--outfile 改成其它的就可以了。结果中得到的 anno.exonic_variant_function为后缀的文件是非常重要的,
$ wc -l WGS-PDAC-sun.anno.exonic_variant_function #查看变异个数
$ less WGS-PDAC-sun.anno.exonic_variant_function #可以看到外显子上的变异
曾得到的结果以及大小如下,看后可以删了,很奇怪我得到的啥都没有。简直fuck!曾说外显子测序可以找到十多万变异,但是在外显子上的只有两三万左右,再根据质量值一过滤就没有两三万了。
外显子变异的解释

至于nonsynonymous是否会导致严重后果可以通过SIFT,PolyPhen-2以及PROVEAN软件来预测SNV对蛋白质功能的影响程度,PROVEAN软件可预测Indel对蛋白质功能的影响。也可以通过dbNSFP数据库注释这些变异对蛋白功能的影响,该数据库现在已经更新了,现在可能更大。利用该数据库就可以避免使用这些软件,该数据库在谷歌云上是比较难下的。

2. 基于区域的注释:略
3. 基于数据库的过滤:略
补充作业
使用 Variant Effect Predictor 对所有遗传变异进行注释。过滤掉 dbSNP 数据库和千人基因组计划数据库中已知的 SNP(可以通过ANNOVAR下载这些数据库)。
过滤
过滤主要是变异位点的过滤
使用 GATK的 Joint Calling , 过滤参考:https://mp.weixin.qq.com/s/W8Vfv1WmW6M7U0tIcPtlng
曾是分析的三个人,父亲、母亲、儿子,通过比较父母和儿子,找父母都没有而儿子却有的变异位点,可能是是出生的时候受环境影响产生的突变。找到这种变异很有意义,首先能增加人类的知识库,将来有其它的性状可以快速定位,同时在小孩子早期的时候还可以进行基因治疗。
下游分析需要自己去学,教程如下
《GATK4的gvcf流程》 https://mp.weixin.qq.com/s?src=11×tamp=1588379895&ver=2313&signature=OCTAnBoTYWu1izLX0hBTsWTX-VeehzWSmG4P64U4cAkbZDsikSv7wBXqE2Cow9yxHJr75aZtGrD-POtLm1UpRwp8ZdQtbkzgKdxdfKmNTsuVCDm3d0iPDnHIcJ1p&new=1
《新鲜出炉的GATK4培训教材全套PPT,赶快下载学习吧》https://mp.weixin.qq.com/s?src=11×tamp=1588379972&ver=2313&signature=OCTAnBoTYWu1izLX0hBTsWTX-VeehzWSmG4P64U4cA-xVhGy15-LNhFyUymVEgXgOcMj4KLrRMc1lTx1UNjiZ3PweMl*tJJ5W0V7WMGPxDJCaAqmBKXVXGcySjJok2tK&new=1
肿瘤外显子和家系外显子
《把vcf文件转换为maf格式,肿瘤外显子上游分析教程到此为止》https://mp.weixin.qq.com/s?src=11×tamp=1588380272&ver=2313&signature=OCTAnBoTYWu1izLX0hBTsWTX-VeehzWSmG4P64U4cA-JaNFGfTCvVTwLLXO62PkKe4Ne3Xot4HEN*GOJIwHrUdN2iq7GJn43SQx-CBUHNDWvR7Ex4yzqdMq-i1GQQUmQ&new=1
参考文章
一定要看的几篇文章
曾说不缺教程,他写了一万多篇教程,生信用到的所有软件都有教程。
《(全)WES(全外显子测序)分析流程》 https://www.jianshu.com/p/beec97dfb05d
《一个全基因组重测序分析实战》 #这里面还讲了构建索引,bwa mem使用的参数也是从这里面拷贝的
《基因组变异分析——GATK安装使用》
《GATK4操作文档》http://www.bio-info-trainee.com/3144.html
《GATK使用注意事项》http://www.bio-info-trainee.com/838.html
《WES 分析(从下载数据到IGV)》 https://www.jianshu.com/p/4519d2e64a49
《ANNOVAR 软件用法还可以更复杂》
《ANNOVAR软件使用》https://www.jianshu.com/p/c8cea664469b
《从零开始完整学习全基因组测序数据分析:第4节构建WGS主流程》这个极其好!!!
《IGV加载很久很烦人?三步帮你解决!》
《使用 Travis 将 GitHub 文件上传传至服务器》
《肿瘤外显子数据处理系列教程�(一)读文献并且下载测序数据》以及一系列的文章
《1 wes相关软件下载与安装并添加环境变量》https://www.jianshu.com/p/e226fcde9a13
《2 下载GATK需要的参考基因组文件》https://www.jianshu.com/p/155882b3005a
《3 wes测序质量的控制》 https://www.jianshu.com/p/830646c00cf2
《3_0_4 要理解并会用的几个脚本》https://www.jianshu.com/p/54e56911dbb5
《4 比对到参考基因组输出bam文件》 https://www.jianshu.com/p/26070125abf6
《5 一步法找基因变异流程》 https://www.jianshu.com/p/7752a6633d1a
《6 GATK4完整流程》https://www.jianshu.com/p/65705f6652ba
《生物信息学最佳实践–基础篇》
整理好并进行渲染,在我的公众号进行发表!
-
《给学徒的WES数据分析流程》 https://www.jianshu.com/p/49d035b121b8 ↩ ↩
-
《全外显子测序(wes)数据分析详细流程(小样本)》https://www.jianshu.com/p/4b677654be15 ↩ ↩ ↩
-
《A beginners guide to SNP calling from high-throughput DNA-sequencing data》 ↩
-
《应用全外显子组测序筛查一高原肺水肿家系易感基因》 ↩
-
《全外显子组测序检测视网膜色素变性家系的致病基因》 ↩
-
上海晶能生物-最新外显子组测序数据分析报告 https://www.docin.com/p-1158054405.html ↩
-
千年基因外显子组测序应用手册.pdf https://www.renrendoc.com/p-41111104.html ↩
-
【直播】我的基因组25:用bcftools来call variation ↩
-
《各种NGS组学数据分析异同点视频讲解》 ↩