镜像设置
①在Rstudio的程序中设置
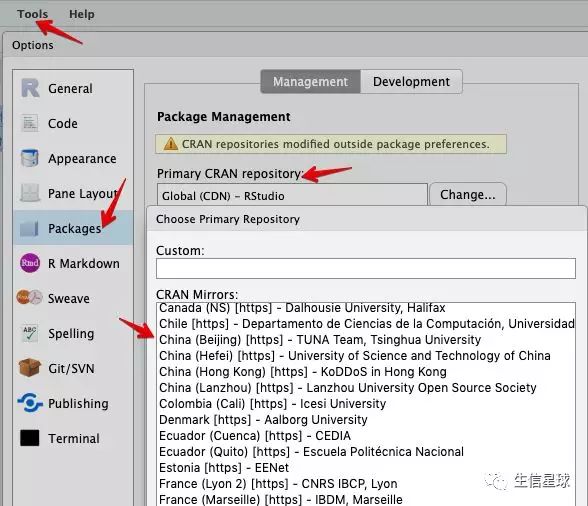
此处设置的是CRAN的镜像,生信也常用到Bioconductor内的包,需要从新设置。
②代码设置
# options函数就是设置R运行过程中的一些选项设置
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/")) #对应清华源
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/") #对应中科大源
# 当然可以换成其他地区的镜像
#检测镜像
options()$repos
options()$BioC_mirror
③直接设定R的配置文件
Rstudio有两个配置文件:
.Renviron设置R的环境变量而.Rprofile是一个代码文件,启动时找到这个文件,就在启动Rstudio时先运行一遍。
源【生信星球】
先打开Rstudio,运行file.edit('~/.Rprofile')编辑文件。
再添加代码:
# options函数就是设置R运行过程中的一些选项设置
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/")) #对应清华源
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/") #对应中科大源
# 当然可以换成其他地区的镜像
保存并重启
#检测镜像
options()$repos
options()$BioC_mirror
安装R包
有两种安装R包命令:
install.packages()和BiocManager::install()
加载R包
一般使用library()命令,require()也可以加载,但严格来说是在尝试加载,一般用来运行为:
if(! require("dplyr")) install.packages("dplyr")
#表示试加载dplyr包,如果没有则安装dplyr包
library("dplyr")
学习dplyr包
dplyr包:主要用来整理数据的工具
准备工作:
#设置镜像
#options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
#options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
#检测镜像
options()$repos
options()$BioC_mirror
#安装和加载包
install.packages("dplyr")
library(dplyr)
#使用dplyr包处理数据前,建议先将数据集转换为tbl对象。
#tbl_df(data)
dplyr包几个基础函数
① mutate()新增列
其语法为select(.data,…)
mutate()返回的结果集会保留原有变量。
② select()按列筛选
select()用列名作参数来选择子数据集。筛选变量时,可与以下函数结合:starts_with,ends_with,contains,matches,one_of,num_range和everything等。
select()也可用于给某列重命名,同rename(),但select()只保留参数中给定的列,rename()保留所有的列,只对给定的列重新命名。
③ filter()筛选行
filter('文件名','列名' == " "
#从'文件名'中筛选满足列名为“ ”值
④ arrange()排序
默认是按照升序排序,对列名加 desc() 可实现倒序排序。
arrange(test, desc(‘列名’))#用desc从大到小
⑤ summarise()汇总
对数据进行汇总操作,结合group_by使用实用性强
group_by用于对数据集按照给定变量分组,返回分组后的数据集。
summarise(group_by(test, Species),mean(Sepal.Length), sd(Sepal.Length))
#test 文件名,Species和Sepal.Length 是列名,mean表示平均值,sd为标准差
⑥distinct去重复值
distinct(.data, ..., .keep_all = FALSE)
其他函数可查看必学包之dplyr包
dplyr包两个实用技能
1.管道操作 %>%
%>%快捷键为(cmd/ctr + shift + M)
tidyverse包是一个汇总包,用于数据筛选、转换、可视化等。
其核心包有ggplot、readr、tibble、purrr、 tidyr 、dplyr、ggplot、forcats 和stringr8个。
用管道符号可加载任意一个tidyverse包。
具体见R语言中管道操作 %>%, %T>%, %$% 和 %%
2.count()统计某列的unique值
类似与Excel内的函数count值。
dplyr处理关系数据
#先避免有因子
options(stringsAsFactors = F)
1.inner_join()取交集
2.left_join()从右侧找左侧值
3.full_join()取并集,自动补NA值
4.semi_join()返回能够与y表匹配的x表的所有记录
5.anti_join()返回无法与y表匹配的x表的所有记录
6.bind_cols()同行数数据框合并;bind_rows()同列数数据框合并
完!
附:本文涉及的例子
#设置镜像
#options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
#options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
#检测镜像
options()$repos
options()$BioC_mirror
#安装和加载包
install.packages("dplyr")
library(dplyr)
#调取内置数据集iris的简化版
test <- iris[c(1:2,51:52,101:102),]
#使用dplyr包处理数据前,建议先将数据集转换为tbl对象。
#test<-tbl_df(test)
#mutate(),新增列
mutate(test, new = Sepal.Length * Sepal.Width)
#select()筛选,结合one_of使用
vars <- c("Petal.Length", "Petal.Width")
select(test, one_of(vars))
#与rename的区别
rename(test, petal_length = Petal.Length)
select(test, petal_length = Petal.Length)
#filter()`筛选行
filter(test, Species == "setosa")
#排序
arrange(test, desc(Sepal.Length))
#汇总
summarise(group_by(test, Species),mean(Sepal.Length), sd(Sepal.Length))
#去重
distinct(test, Petal.Length, keep_all = FALSE)
#dplyr处理关系数据
#先避免有因子
options(stringsAsFactors = F)
test1 <- data.frame(x = c('b','e','f','x'),
z = c("A","B","C",'D'),
stringsAsFactors = F)
test2 <- data.frame(x = c('a','b','c','d','e','f'),
y = c(1,2,3,4,5,6),
stringsAsFactors = F)
inner_join(test1, test2, by = "x")
left_join(test1, test2, by = 'x')
left_join(test2, test1, by = 'x')
full_join( test1, test2, by = 'x')
semi_join(x = test1, y = test2, by = 'x')
anti_join(x = test2, y = test1, by = 'x')
#合并
test3 <- data.frame(x = c(1,2,3,4), y = c(10,20,30,40))
test4 <- data.frame(x = c(5,6), y = c(50,60))
test5 <- data.frame(z = c(100,200,300,400))
bind_rows(test3, test4)
bind_cols(test3, test5)