本节,我们演示了在Seurat对象、singlecellexper对象和anndata对象之间进行转换的能力。以建立Seurat与其他单细胞数据分析工具之间的链接。
# install scater https://bioconductor.org/packages/release/bioc/html/scater.html
library(scater)
# install loomR from GitHub using the remotes package remotes::install_github(repo =
# 'mojaveazure/loomR', ref = 'develop')
library(loomR)
library(Seurat)
library(monocle)
Seurat to monocle
用 Seurat 3 的Seurat::as.CellDataSet()函数可以直接将Seurat对象转化为monocle2的对象,进行monocle2的拟时分析。
?Seurat::as.CellDataSet()
monocle to Seurat
那,monocle分析之后又想用seurat的功能怎么办呢?monocle的对象也是可以转化为Seurat的。在这之前,让我们来看看monocle的数据格式:
一般monocle构建CDS需要3个矩阵:expr.matrix、pd、fd
- expr.matrix :基因-细胞表达矩阵
- pd :细胞-细胞特征注释矩阵
- fd :基因-基因特征注释矩阵
而创建这个seurat对象需要什么呢?
?Seurat::CreateSeuratObject
CreateSeuratObject(
counts, # 一个基因-细胞表达矩阵 (对应monocle的expr.matrix)
project = "SeuratProject", # 项目名称可以以您的心情写
assay = "RNA", # 一般count矩阵说就是RNA
min.cells = 0, # cells 过滤条件
min.features = 0, # 基因过滤条件
names.field = 1, # 细胞名称格式(一般不用,geo下载的数据需要注意了)
names.delim = "_", # 细胞名称分隔符(一般不用)
meta.data = NULL #细胞-细胞特征注释矩阵
)
所以我们可以用monocle的 expr.matrix 和pd 来构建seurat的对象。
converting-tofrom-singlecellexperiment
SingleCellExperiment是由Davide Risso、Aaron Lun和Keegan Korthauer创建的用于存储单细胞实验数据的类,并被许多生物导体分析包使用。在这里,我们演示了将3k PBMC教程中生成的Seurat对象转换为与Davis McCarthy的scater包一起使用的SingleCellExperiment对象。
# download from satija lab https://www.dropbox.com/s/kwd3kcxkmpzqg6w/pbmc3k_final.rds?dl=0
pbmc <- readRDS(file = "../data/pbmc3k_final.rds")
pbmc.sce <- as.SingleCellExperiment(pbmc)
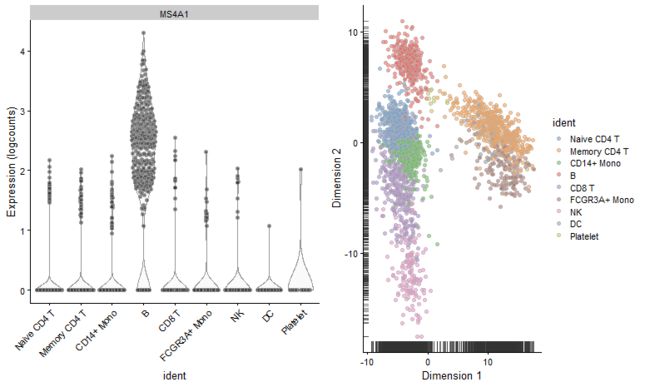
p1 <- plotExpression(pbmc.sce, features = "MS4A1", x = "ident") + theme(axis.text.x = element_text(angle = 45,
hjust = 1))
p2 <- plotPCA(pbmc.sce, colour_by = "ident")
CombinePlots(plots = list(p1, p2))
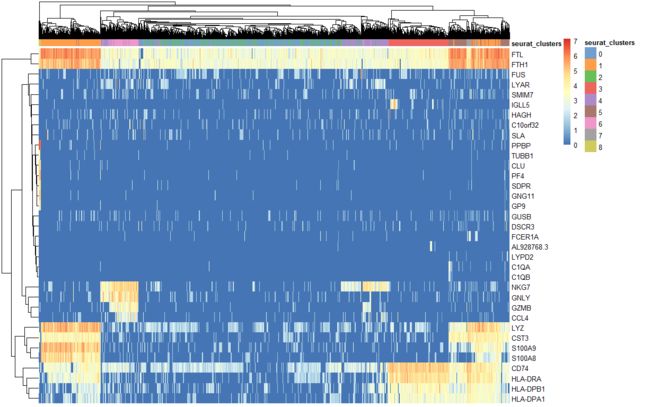
?pheatmap
head([email protected])
plotHeatmap(pbmc.sce,features = VariableFeatures(object = pbmc) ,show_colnames=F,colour_columns_by='seurat_clusters')#,cluster_cols=F
pbmc.sce <- calculateQCMetrics(pbmc.sce)
fontsize <- theme(axis.text=element_text(size=12), axis.title=element_text(size=16))
plotHighestExprs(pbmc.sce, n=20) + fontsize

Seurat还允许从SingleCellExperiment对象转换为Seurat对象;我们通过从Martin Hemberg的团队维护的存储库下载的一些公共数据来演示这一点。
# download from hemberg lab
# https://scrnaseq-public-datasets.s3.amazonaws.com/scater-objects/manno_human.rds
manno <- readRDS(file = "D:\\Users\\Administrator\\Desktop\\Novo周运来\\SingleCell\\scrna_tools/manno_human.rds")
manno <- runPCA(manno)
manno.seurat <- as.Seurat(manno, counts = "counts", data = "logcounts")
# gives the same results; but omits defaults provided in the last line
manno.seurat <- as.Seurat(manno)
Idents(manno.seurat) <- "cell_type1"
p1 <- DimPlot(manno.seurat, reduction = "PCA", group.by = "Source") + NoLegend()
p2 <- RidgePlot(manno.seurat, features = "ACTB", group.by = "Source")
CombinePlots(plots = list(p1, p2))

converting-tofrom-loom
loom是一个文件结构强加于HDF5文件,由斯坦林纳森的小组设计。它的设计是为了有效地描述大型单细胞基因组数据集。loop格式的详细信息,请参阅http://linnarssonlab.org/loompy/format/index.html
pbmc.loom <- as.loom(pbmc, filename = "pbmc3k.loom", verbose = FALSE)
Transposing input data: loom file will show input columns (cells) as rows and input rows (features) as columns
This is to maintain compatibility with other loom tools
Adding: CellID
Adding: Gene
Adding: vst_mean
Adding: vst_variance
Adding: vst_variance_expected
Adding: vst_variance_standardized
Adding: vst_variable
Adding: Selected
Adding: orig_ident
Adding: nCount_RNA
Adding: nFeature_RNA
Adding: percent_mt
Adding: RNA_snn_res_0_5
Adding: seurat_clusters
Adding: S_Score
Adding: G2M_Score
Adding: Phase
Adding: ClusterID
Adding: ClusterName
> pbmc.loom
Class: loom
Filename: D:\Documents\pbmc3k.loom
Access type: H5F_ACC_RDWR
Attributes: version, chunks, LOOM_SPEC_VERSION, assay, last_modified
Listing:
name obj_type dataset.dims dataset.type_class
col_attrs H5I_GROUP
col_graphs H5I_GROUP
layers H5I_GROUP
matrix H5I_DATASET 2638 x 13714 H5T_FLOAT
row_attrs H5I_GROUP
row_graphs H5I_GROUP
# Always remember to close loom files when done
pbmc.loom$close_all()
当然了,Seurat也支持loom格式到seurat的转化。我们在Linnarson实验室制作的小鼠大脑图谱Mouse Brain Atlas
的一个子集中进行演示。(我还是用Seurat生成的pbmc3k.loom来做演示吧……)
> pbmc <- connect(filename = "pbmc3k.loom", mode = "r")
> pbmc
Class: loom
Filename: D:\Documents\pbmc3k.loom
Access type: H5F_ACC_RDONLY
Attributes: version, chunks, LOOM_SPEC_VERSION, assay, last_modified
Listing:
name obj_type dataset.dims dataset.type_class
col_attrs H5I_GROUP
col_graphs H5I_GROUP
layers H5I_GROUP
matrix H5I_DATASET 2638 x 13714 H5T_FLOAT
row_attrs H5I_GROUP
row_graphs H5I_GROUP
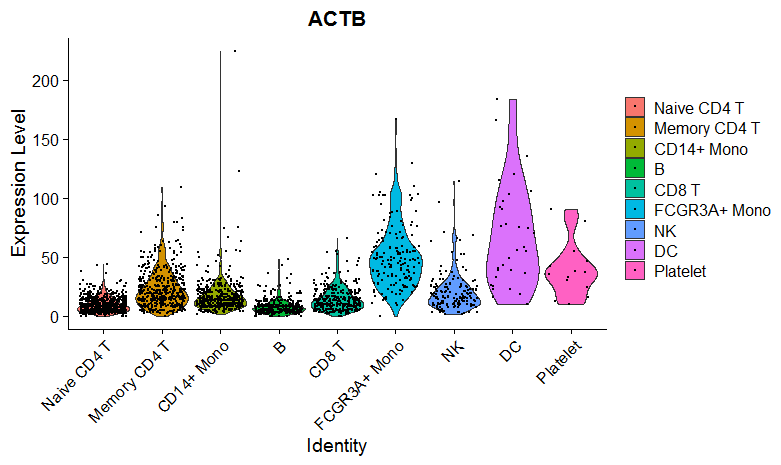
pbmc <- as.Seurat(pbmc)
VlnPlot(pbmc, features ="ACTB" , ncol = 2, pt.size = 0.1)
# Always remember to close loom files when done
l6.immune$close_all()
converting-tofrom-anndata
AnnData提供了一个由Alex Wolf和Philipp Angerer创建的Python类,它可以用来存储单细胞数据。这种数据格式也用于存储在他们的Scanpy包中,我们现在支持互操作性。Seurat有一个新的函数ReadH5AD,用于从AnnData使用的H5AD文件中读取数据。
# download from satija lab https://www.dropbox.com/s/ngs3p8n2i8y33hj/pbmc3k.h5ad?dl=0
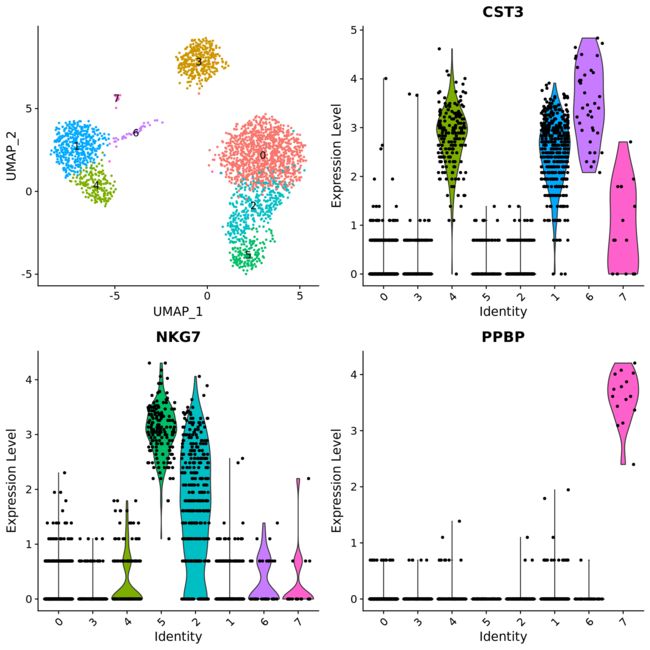
pbmc3k <- ReadH5AD(file = "../data/pbmc3k.h5ad")
Idents(pbmc3k) <- "louvain"
p1 <- DimPlot(pbmc3k, label = TRUE) + NoLegend()
p2 <- VlnPlot(pbmc3k, features = c("CST3", "NKG7", "PPBP"), combine = FALSE)
CombinePlots(plots = c(list(p1), p2), ncol = 2, legend = "none")
We currently do not support direct Seurat → AnnData conversion. We do have a work around by allowing users to write out to a loom file with as.loom, then reading the loom file in Python with Scanpy/AnnData
conversion_vignette