我们已经使用Tidyr将数据进行了变形,使其符合Tidy data的原则。而在对数据进行正式的统计分析之前,通常还需要进行一些必要的数据处理,如:
- 改变结构:排序,修改列名
- 按行操作:过滤,选取,抽样
- 按列操作:筛选特定列
- 统计、分组统计:最大值,最小值等
要完成以上这些分析,我们可以使用R中非常强大的一个包dplyr,通过几个简单的命令,即可完成我们想要的分析。
1.dplyr包的安装和加载
# 安装dplyr包
>install.packages("dplyr")
# 加载dplyr包
>library(dplyr)
2.使用dplyr进行数据管理
(1)排序:arrange
在使用tidyr对数据进行处理后,如下:
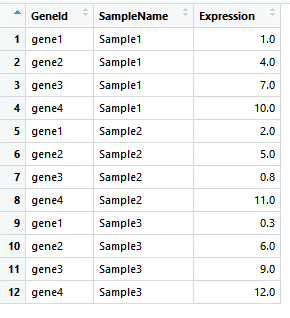
image
在上面的图表中,是以SampleName进行排序的,我们可以以GeneId进行排序:
# arrange(数据框名,要排序的列名)
gene_exp_GeneId <- arrange(gene_exp_tidy, GeneId)
按GeneId排序后的效果如下:

image
如果我们想要在GeneId排序的基础上,再以Expression排序:
gene_exp_GeneId_Exp <- arrange(gene_exp_tidy, GeneId, Expression)
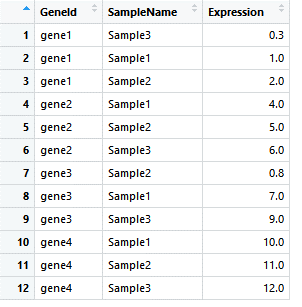
image
默认条件下,是以升序排列的,如果我们想要以降序排列,只需在需要降序排列的列名前添加desc即可:
# 降序排列
gene_exp_GeneId_descExp <- arrange(gene_exp_tidy, GeneId, desc(Expression))
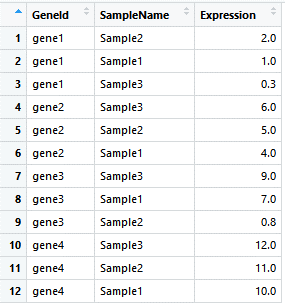
image
(2)按行(观测)筛选:filter
筛选出表达量大于1的观测:
# filter(数据框名,筛选条件)
filter(gene_exp_tidy, Expression > 1)

image
筛选出表达量大于1,并且基因名为gene1的观测:
filter(gene_exp_tidy, Expression > 1, GeneId == "gene1")

image
(3)按列筛选:select
select函数用于选择需要展示的列:
# 只展示SampleName和Expression列
select(gene_exp_tidy, SampleName, Expression)

image
使用select函数,我们还可以进行模糊的筛选:
# 筛选所有列名中包含"me"的列
select(gene_exp_tidy, contains("me"))

image
# 展示所有列名中,以G开始的列
select(gene_exp_tidy, starts_with("G"))

image
(4)管道操作符:%>%
我们对数据的筛选和排序操作,可以使用管道符连接起来,一步即可完成:
# 先筛选出表达量大于1的观测,再按GeneId升序排列,同时按Expression降序排列
filter(gene_exp_tidy, Expression >1) %>% arrange(GeneId, desc(Expression))
# 说明:使用管道符连接时,第一个操作的输出结果会成为第二个操作的输入,因此在arrange中不再需要指定数据框的名称

image
(5)去除重复:distinct
使用distinct函数,我们可以去除数据框中有重复的行:
# 去除GeneId中有重复的行
distinct(gene_exp_tidy, GeneId)
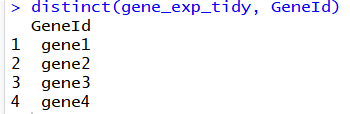
image
(6)添加新变量:mutate
mutate函数用于根据已有的变量,产生一个新的变量:
# 产生一个表达量是现有表达量10倍的变量
mutate(gene_exp_tidy, "10Exp" = Expression * 10)

image
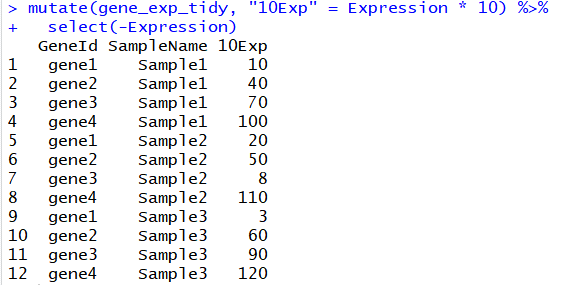
# 得到表达量变为10倍的变量后,去除原来的Express列
mutate(gene_exp_tidy, "10Exp" = Expression * 10) %>%
select(-Expression)
image
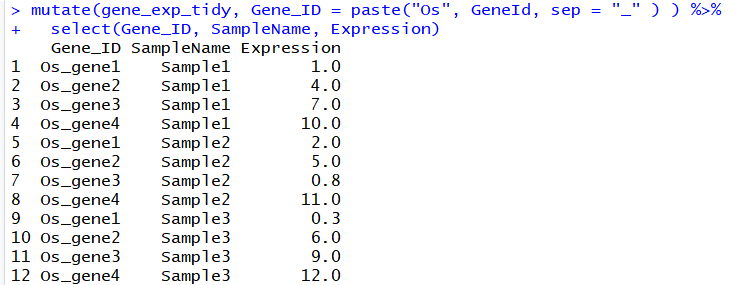
# 在每一个GeneId前面,添加Os字符,用于说明是水稻的基因,并去除原来的GeneId列
# paste函数用来连接字符
mutate(gene_exp_tidy, Gene_ID = paste("Os", GeneId, sep = "_" ) ) %>%
select(Gene_ID, SampleName, Expression)
image
(7)分组统计:group_by %>% summarise
使用summarise函数,可以对数据进行简单的统计:
# 计算Expressin的平均值
summarise(gene_exp_tidy, avg = mean(Expression))

image
先对数据进行分组,再进行统计:
# 先以GeneId进行分组,再计算平均值
gene_exp_tidy %>%
group_by(GeneId)%>%
summarise(avg = mean(Expression))

image
# 计算每个基因在三个样品中表达量的标准差sd
gene_exp_tidy %>%
group_by(GeneId)%>%
summarise(sd = sd(Expression))

image