Single-Cell RNA-Seq Technologies and Related Computational Data Analysis
单细胞RNA测序(scRNA-seq)技术允许在单细胞分辨率下解析基因表达,这极大地改变了转录组学研究。目前已经开发了大量的scRNA-seq protocol,这些protocol各有特点,各有优缺点。由于技术限制和生物因素,scRNA-seq数据比 bulk RNA-seq数据更嘈杂、更复杂。scRNA-seq数据的高度可变性给数据分析带来了计算方面的挑战。虽然越来越多的生物信息学方法被提出用于分析和解释scRNA-seq数据,但需要新的算法来确保结果的准确性和再现性。在本文中,我们概述现有的一些protocol和scRNA-seq单细胞分离技术,并讨论不同方法scRNA-seq数据分析包括质量控制、read mapping, 基因表达量化、批次效应校正,规范化,imputation,降维,特征选择,细胞聚类、轨迹推理,差异表达calling,可变剪接,等位基因的表达,基因调控网络重构。此外,我们概述了scRNA-seq技术的发展和应用前景。
Introduction:
在过去的十年中,Bulk RNA-seq技术被广泛应用于研究群体水平上的基因表达模式。单细胞RNA测序(scRNA-seq)的出现为探索单细胞水平的基因表达谱提供了前所未有的机会。目前,scRNA-seq已经成为研究细胞异质性和早期胚胎发育等关键生物学问题的良好选择,因为bulk RNA-seq主要反映了数千个细胞的平均基因表达。近年来,scRNA-seq已被应用于不同的物种,特别是不同的人体组织(包括正常和癌症),这些研究揭示了有意义的细胞间基因表达变异.近年来,随着测序技术的不断创新,一些不同的scRNA-seq protocal被提出,极大地促进了对单细胞动态基因表达的理解。其中一种是高效的LCM-seq (Nichterwitz et al., 2016),它结合了激光捕获显微镜(LCM)和Smart-seq2 (Picelli et al., 2013),用于单细胞转录组学,不用分离组织 。目前可用的scRNA-seq protocol 根据捕获的转录覆盖率主要可分为两类:(i)全长转录测序方法[如Smart-seq2 (Picelli et al., 2013)、MATQ-seq (Sheng et al., 2017)和SUPeR-seq (Fan X. et al., 2015)];和(ii) 3 -端, Drop-seq (Macosko et al. ,2015), Seq-Well (Gierahn et al., 2017), Chromium (Zheng et al., 2017), and DroNC-seq (Habib et al., 2017)] or 5 -end[如STRT-seq (Islam et al., 2011, 2012)]转录测序技术。每个scRNA-seq协议都有其优缺点,导致不同的scRNA-seq方法具有不同的特点和不同的性能(Ziegenhain et al., 2017)。在进行单细胞转录组研究时,考虑到研究目标和测序成本之间的平衡,可能需要使用特定的scRNA-seq技术。
思考:这些protocol中为什么会区分3'-end 和5'-end?
由于起始量少,所以scRNA-seq具有捕获效率低、漏检率高的局限性。与 Bulk RNA-seq相比,scRNA-seq产生的数据更嘈杂、更多变。技术噪声和生物变异(如随机转录)对scRNA-seq数据的计算分析提出了实质性的挑战。已经设计了各种工具来进行不同的bulk RNA-seq数据分析,但其中许多方法不能直接应用于scRNA-seq数据。除了short-read mapping,几乎所有的数据分析(如差异表达、细胞聚类和基因调控网络推断)在方法上都存在一定的差异。由于高技术噪声,质量控制(QC)是识别和去除低质量scRNA-seq数据的关键,以获得可靠的和可重复的结果。此外,一些分析包括可变剪接(AS, Alternative Splicing)检测、等位基因表达勘探和rna编辑识别不适合scRNA-seq 3'或5'端测序 protocol, 但这些分析方法可以适用于whole-transcript scRNA-seq protocol 产生的数据。另一方面,越来越多的工具是专门为scRNA-seq数据分析提出,每种方法都有自己的优点和缺点。因此,为了有效地处理高可变性的scRNA-seq数据,要注意选择适当的分析方法。
本文旨在总结和讨论当前可用的scRNA-seq技术和各种数据分析方法。我们首先介绍了独特的单细胞分离方法和各种scRNA-seq技术,这是近年来发展起来的。然后,我们重点分析了scRNA-seq数据,并强调了Bulk RNA-seq和scRNA-seq数据之间的分析差异。考虑到scRNA-seq数据的高技术噪声和复杂性,我们还对如何选择合适的工具来分析scRNA-seq数据并确保结果的再现性提出了建议。
单细胞分离(Isolation of Single Cells)
尽管对scRNA-seq来说,捕获效率是一个很大的挑战,但是scRNA-seq的第一步是分离单个细胞(图1)。目前,分离单个细胞有几种不同的方法,包括有限稀释法(Limiting dilution technique)、显微操作法(micromanipulation)、流式细胞分选(FACS)、激光捕获显微分离(LCM)和微流控(microfluidics)。有限稀释技术(Limiting dilution technique)是利用移液管稀释分离细胞,这种方法的主要缺点是效率低下。显微操作是一种经典的方法,用于从少量细胞样本中提取细胞,如早期胚胎或未培养的微生物,而这种技术是耗时和低通量的。流式细胞仪广泛用于分离单个细胞,在悬浮状态下需要较大的起始体积(>10,000个细胞)。LCM是一种先进的技术,利用计算机辅助的激光系统将单个细胞从固体组织中分离出来。微流控技术以其样品消耗量低、流体控制精确、分析成本低等特点,越来越受到人们的重视。这些单细胞分离方案具有各自的优点,在捕获效率和目标细胞纯度方面表现出明显的性能。

现有scRNA-seq技术(Currently Available ScRNA-Seq Technologies)
至今,许多scRNA-seq技术被提出用于单细胞转录组研究(表1)。Tang等人(2009)发表了第一个scRNA-seq方法,随后又开发了许多其他scRNA-seq方法。这些scRNA-seq技术至少在以下一个方面不同:(i) 细胞分离; (ii)细胞裂解; (iii) 反转录;(iv) 扩增;(v) 转录本覆盖度;(vi) 链特异性; (vii) UMI(独特的分子标识符,可用于检测和量化独特转录本的分子标记)可用性。这些scRNA-seq方法显著区别之一是,他们中的一些人可以产生全长(或几乎全长)转录测序数据(例如,Smart-seq2 SUPeR-seq, MATQ-seq),而其他人只捕获和序列的3' 端(如Drop-seq Seq-Well DroNC-seq, SPLiT-seq(罗森博格et al ., 2018))或 5' 端(例如,STRT-seq)记录(表1),不同的scRNA-seq protocol 可能拥有不同的优势和劣势,一些发表的文章已经详细比较了其中的一部分软件。之前的研究表明,Smart-seq2可以检测到比其他scRNA-seq技术更多的表达基因,包括cell-seq2、mas-seq、Smart-seq 和 Drop-seq protocol。最近,Sheng等人(2017)表明,另一种全长转录测序方法MATQ-seq在检测低丰度基因方面可以优于Smart-seq2。
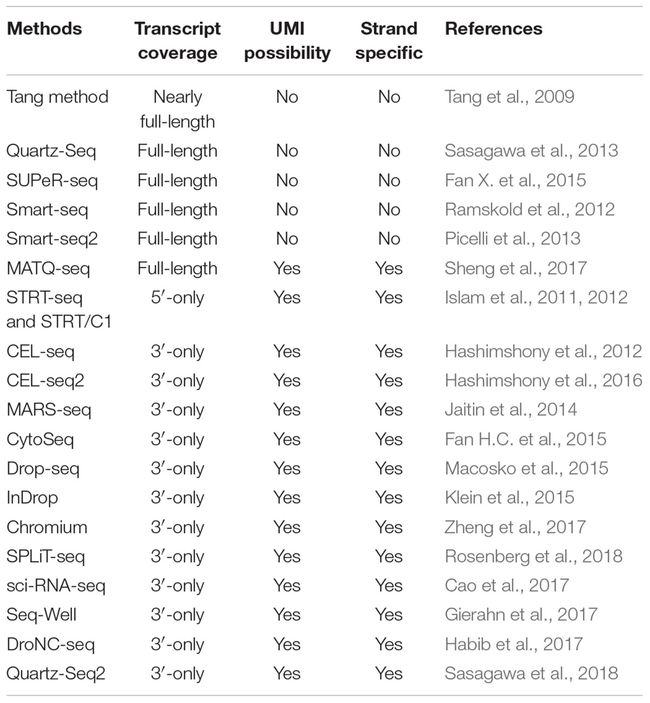
与3’端或5’端计数方法相比,全长scRNA-seq方法在亚型使用分析、等位基因表达检测、RNA编辑鉴定等方面具有无可比拟的优势。此外,对于某些低表达基因/转录本的检测,全长scRNA-seq方法可能优于3’测序方法。值得注意的是,基于droplet-based的技术[例如, Drop-seq (Macosko et al., 2015)、InDrop (Klein et al., 2015)和Chromium (Zheng et al., 2017)]通常可以提供更大的细胞通量,而且与全转录scRNA-seq相比,每个细胞的测序成本更低。因此,基于droplet-based的方案适合生成大量细胞来识别复杂组织或肿瘤样本的细胞亚群。
值得注意的是,有几种scRNA-seq技术可以同时捕获polyA阳性和polyA阴性的 RNAs,如SUPeR-seq (Fan X. et al., 2015)和MATQ-seq (Sheng et al., 2017)。这些protocol对于长链非编码rna (lncRNAs)和环状rna (circRNAs)的测序非常有用。大量研究表明,lncRNAs和circRNAs在细胞的多种生物学过程中发挥重要作用,可能是癌症的重要生物标志物。因此,这种scRNA-seq方法可以提供前所未有的机会,在单细胞水平上全面探索蛋白质编码和非编码rna的表达动态。
与传统的Bulk RNA-seq技术相比,scRNA-seq protocol 存在较大的技术差异。为了评估不同细胞间的技术差异,spikein[如 External RNA Control Consortium (ERCC) controls (External, 2005)]和UMIs被广泛应用于相应的scRNA-seq方法中。RNA spike-in 是RNA转录本(具有已知的序列和数量),用于校准RNA杂交测定的测量结果,如RNA-seq,而UMIs在理论上可以用于估算绝对分子数。值得注意的是,ERCC和UMIs并不适用于所有的scRNA-seq技术,因为它们之间存在固有的protocol差异。spikein用于Smart-seq2和SUPeR-seq等方法,但与基于droplet-based 方法不兼容,而UMIs通常用于3' 端测序技术[如Drop-seq (Macosko et al., 2015)、InDrop (Klein et al., 2015)和MARS-seq (Jaitin et al., 2014)]。因此,用户可以根据技术特性和优点、需要测序的细胞数和成本考虑来选择合适的scRNA-seq方法。
思考:spike-in和UMI的作用是什么?
单细胞数据的比对和质控(Read Alignment and Expression Quantification of ScRNA-Seq Data)
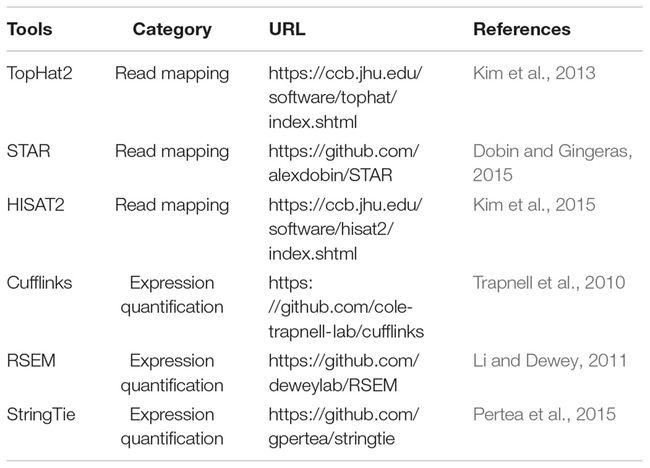
reads的比对率是scRNA-seq数据整体质量的重要指标。由于scRNA-seq和bulk RNA-seq技术通常都是将转录本序列转换为read来生成fastq格式的原始数据,所以这两种类型的RNA-seq数据在read 比对方面没有区别。最初为bulk RNA-seq开发的比对工具也适用于scRNA-seq数据。为比对RNA-seq数据,已经设计了许多比对软件,这在之前已经广泛讨论过。通常,比对算法主要分为两类:基于空间种子索引的和基于Burrows-Wheeler transform (BWT)的(Li and Homer, 2010)。目前流行的比对软件:TopHat2,STAR,和HISAT 在速度和准确性上都表现很好,并且他们可以有效地数十亿read 比对到参考基因组或转录组(表2)。STAR是基于suffix-array方法,比TopHat2更快,但它比对时需要一个巨大的内存大小(28 g用于人类基因组)。Engstrom等人系统地评估了26种read比对protocol(不包括HISAT),发现不同的比对工具有不同的优缺点,有些程序的比对速度更快,但拼接连接检测的准确性更低。HISAT是基于BWT和Ferragina-Manzini (FM)方法开发的。Kim等人(2015)指出,HISAT是目前最快的工具,可以达到与其他可用对准器相同或更好的精度。
数据质控(Quality Control of ScRNA-Seq Data)
scRNA-seq的局限性包括转录本覆盖的偏差、低捕获效率和测序覆盖度低,导致scRNA-seq数据的技术噪声水平高于bulk RNA-seq数据。即使对一些很灵敏的scRNA-seq protocol,
也是个很常见的现象,一些特定的转录本无法被检测到,这个术语叫dropout events。通常,scRNA-seq实验可以从破损、死亡或与多个细胞混合的细胞中生成部分低质量的数据。
这些低质量的细胞将阻碍下游的分析,并可能导致数据的误读。因此,对scRNA-seq数据的QC质量控制是识别和去除低质量细胞的关键。
为了排除scRNA-seq中的低质量细胞,在细胞捕获步骤中应密切注意避免多细胞或死亡细胞。测序后,需要进行一系列的QC分析来剔除来自低质量细胞的数据。由于测序深度不足可能导致大量低表达和中度表达的基因丢失,因此这些样本只包含少量的reads,应该首先丢弃。然后,可以使用最初为bulk RNA-seq数据QC开发的工具(如FastQC)来检查scRNA-seq数据的测序质量。此外,在读取比对之后,应该删除比对率非常低的样本,因为它们包含大量未必对上的read,这可能是由于RNA降解造成的。如果在scRNA-seq中使用外源性噪声(如ERCC),则可以对技术噪声进行估计。具有极高比例的比对率比对到spike-in的细胞表明,它们可能在单元格捕获过程中被破坏,应该被删除。细胞质rna通常会丢失,但线粒体rna会保留在受损细胞中,因此,绘制到线粒体基因组的reads比例也有助于识别低质量细胞(Bacher和Kendziorski, 2016)。此外,在每个细胞中可以检测到的表达基因/转录的数量也具有启发性。如果在一个细胞中只能检测到少量的基因,那么这个细胞很可能已经受损或死亡,或者是RNA降解了。考虑到scRNA-seq数据的高噪声,通常采用1 FPKM/RPKM的阈值来定义表达基因。提出了一些scRNA-seq的QC方法包括SinQC 和Scater ,这些工具对scRNA-seq数据的质量控制是有用的。
批次效应校正(Batch Effect Correction)
批次效应是高通量测序实验中常见的技术变异来源。scRNA-seq的创新和成本的降低使得许多研究能够分析大量细胞的转录组。
大规模scRNA-seq数据集可以在不同的时间由不同的操作人员单独生成,也可以在多个实验室使用不同的细胞分离protocol、文库制备方法和或测序平台生成。
这些因素会引入系统错误,混淆技术和生物变异,导致一个批次的基因表达谱与另一个批次的基因表达谱存在系统差异(Leek et al., 2010;(Hicks et al., 2018)。
因此,批次效应是scRNA-seq数据分析的一个主要挑战,它可能掩盖了潜在的生物学特性,并导致虚假的结果。
为了避免不正确的数据整合和解释,批次效应必须在下游分析之前纠正。
由于scRNA-seq与bulk RNA-seq在数据特征上的差异,本文提出了针对bulk RNA-seq的批次校正方法[例如, RUVseq (Risso et al., 2014)和svaseq (Leek, 2014)]可能不适合scRNA-seq。
最近设计了几种方法来缓解scRNA-seq数据中的批处理效应,如MNN(相互最近邻居)(Haghverdi et al., 2018)和kBET (k-最近邻居批处理效应测试)(Buttner et al., 2019)。
MNN使用来自不同批次中最相似细胞的数据校正批次效果。KBET χ2-based方法为量化scRNA-seq数据批处理的影响。
这些针对scRNA-seq数据的特定批处理校正方法可以比针对批次RNA-seq开发的方法表现得更好(Haghverdi et al., 2018;Buttner et al., 2019)。
单细胞数据标准化(Normalization of ScRNA-Seq Data)
为了正确地解释scRNA-seq数据的结果,标准化是通过调整由捕获效率、测序深度、dropouts和其他技术影响引起的不想要的偏差来获得感兴趣信号的重要步骤。由于起始量低(starting material)、实验方案具有挑战性,scRNA-seq的技术噪声是一个明显的问题。scRNA-seq数据的标准化将有利于后续分析,包括细胞亚群识别和差异表达calling。一般来说,normalization可以分为两种不同的类型:样品内标准化和样品间标准化。样品内标准化只要是要消除基因特异性(gene-specific)偏差,这使得基因表达在一个样本内具有可比性(如RPKM/FPKM和TPM)。而样本间归一化则是通过调整样本特异性差异(如测序深度和捕获效率)来实现样本间基因表达的比较。通常,这些简单的标准化策略是基于排序深度或上四分位数。如果在scRNA-seq protocol中使用了spike-in或UMI,则可以根据spike-in /UMIs的perfomance 来进行标准化。
bulk RNA-seq 数据分析中有很多处理样品间标准化的方法。比如DESeq2, TMM(trimmed mean of M values)。DEseq2根据不同样本的read count 来计算比例因子(scaling factor),而TMM则是删除极端的对数倍变化(removes the extreme log fold changes)。但是这种bulk-based标准化方法不能用于scRNA-seq数据,因为scRNA-seq产生大量的零表达值,并且比bulk RNA-seq具有更高的技术变异水平。因此使用bulk RNA-seq 标准化方法可能会导致对低表达基因的scRNA-seq过度校正。当然,有很多处理scRNA-seq标准化的方法,比如SCnorm,SAMstrt 和最近引入的一种反卷积方法,它使用跨细胞池的求和表达式值来进行标准化。SCnorm是基于分位数回归的方法。SAMstrt是基于spike-in。Bacher等(2017)认为,如果用基于bulk RNA-seq开发的传统归一化方法去对scRNA-seq数据做标准化可能会引入artifacts,而SCnorm可以有效地对scRNA-seq数据进行归一化,改善主成分分析(PCA)和差异表达基因的识别。
scRNA-seq数据的imputation(Imputation of ScRNA-Seq Data)
单细胞RNA-seq数据通常包含许多由于原始RNA扩增失败而导致的缺失值或dropouts。(注解: dropouts是指空beats)。dropout 的比例是跟 protocol 相关,而且与每个细胞测序的reads数密切相关。dropout event 增加了细胞间的变异。导致信号对每个基因的影响,模糊了基因-基因关系的检测。由于在scRNA-seq中可能无法检测到大量真正表达的转录本,因此dropout 可能会很大程度上影响下游的分析。Imputation是一种用替代值替换缺失数据(dropouts)的有效方法。虽然已经提出了一些方法来估算bulk RNA-seq数据,但它们并不直接适用于scRNA-seq数据。最近为scRNA-seq开发了几种imputation方法。包括SAVER,MAGIC,ScImpute,DrImpute,AutoImpute。SAVER是一个基于贝叶斯的模型,用于基于umi的scRNA-seq数据来恢复所有基因的真实表达水平。MAGIC通过构建基于马尔可夫亲和度的基因表达图来进行基因表达的计算。ScImpute的开发者认为,SAVER和MAGIC可能会导致未受dropout影响的基因的表达变化,而ScImpute可以利用其他类似细胞中不太可能受dropout影响的相同基因的信息,在不引入新的偏差的情况下计算dropout值。DrImpute是一种基于集群的方法,可以有效地将dropout中的零从真正的零中分离出来。AutoImpute是一种基于自编码的方法,它通过学习scRNA-seq数据的固有分布来寻找(impute)缺失的值。最近,Zhang等人评估了不同的imputation方法,发现这些方法的性能与其模型假设和可扩展性相关。
降维和特征选择(Dimensionality Reduction and Feature Selection)
单细胞RNA测序数据具有高维性,可能涉及数千个基因和大量细胞。降维和特征选择是处理高维数据的两种主要策略。降维方法通常通过最优地保留原始数据的一些关键属性来将数据映射到更低的维度空间中。PCA是一种线性降维算法,它假设数据近似正态分布。T-SNE(T-distributed stochastic neighbor embedding)是一种主要用于高维数据可视化的非线性方法。PCA和t-SNE在大量的的scRNA-seq研究中被广泛应用,以减少数据维度,并将细胞区分为不同的亚群。值得注意的是,PCA不能有效地表示scRNA-seq数据的复杂结构,而t-SNE具有计算时间慢、多次处理同一数据集的嵌入方式不同的局限性。最近,为了减少scRNA-seq数据的维度,专门开发了UMAP (uniform manifold approximation and projection) (Becht et al., 2018)和scvis (Ding et al., 2018)。Becht等人指出,与其他降维方法相比,UMAP提供了最快的运行时间、最高的再现性和最有意义的细胞簇组织(Becht et al., 2018)。
特征选择删除了没有信息的基因,并识别最相关的特征,以减少下游分析中使用的维数。通过执行特征选择来减少基因数量,可以在很大程度上加速大规模scRNA-seq数据的计算。差异表达在bulk RNA-seq实验中是一种广泛应用的特征选择方法,但由于scRNA-seq数据差异表达需要预定的信息或同构的亚群信息,因此很难应用于scRNA-seq数据中。针对scRNA-seq数据设计的无监督特征选择算法可分为以下几组:(i)基于高度可变基因(HVG)的;(2)基于spike-in;和(iii)基于dropout。HVG方法依赖于一种假设,即细胞间表达高度可变的基因是由生物学效应而非技术噪音造成的。HVG方法包括Brennecke等人(2013)提出的算法,以及Seurat实现的FindVariableGenes (FVG) (Satija等人,2015)。基于spikein的方法可以识别出那些表现出显著高于那些表达水平相似的spikein的基因。[如scLVM (Buettner et al., 2015)和BASiCS (Vallejos et al., 2015)],两者对HVG的概念相似。基于Dropout的方法利用scRNA-seq数据的Dropout分布进行特征选择,如M3Drop (Andrews and Hemberg, 2018b)。Andrews和Hemberg证明了他们的M3Drop工具优于现有的基于方差(variance-based)的特征选择方法。
细胞亚群的鉴定(Cell Subpopulation Identification)
scRNA-seq数据分析的一个关键目标是鉴定特定条件或组织内的细胞亚群(不同的细胞群通常是不同的细胞类型),以揭示细胞的异质性。细胞亚群鉴定应在质量控制和scRNA-seq数据标准化后进行,否则可能会引入人为因素。基于先验信息的聚类方法主要可以分为两类。如果使用一组已知marker进行聚类,则这些方法是基于先验信息的。另外,无监督聚类方法也可用于用scRNA-seq数据重新识别细胞群。无监督聚类算法主要分为以下几种类型: (i) k-means;(ii) 层次聚类(hierarchical clustering); (iii) density-based clustering; and (iv) graph-based clustering。K-means是一种快速的方法,它将细胞分配到最近的分类中心,并且它需要预先确定cluster数量。层次聚类可以确定聚类之间的关系,但是它的工作速度通常比k-means慢。基于密度(density-based)的聚类方法需要大量的样本来精确计算密度,通常假设所有的聚类具有相同的密度。graph-based聚类可以看作是基于密度的聚类的一种扩展,可以应用于数百万个细胞。一些为scRNA-seq数据特别设计的聚类方法,如单细胞一致性聚类(single cell consensus clustering, SC3)。和Seurat中的聚类方法(Satija et al ., 2015),它可以更容易的鉴定细胞亚群(表3)。SC3是一种无监督的方法,结合多个聚类方法,在单细胞cluster中具有精度高和稳健性。Seurat主要基于共享近邻(shared nearest neighbor, SNN)聚类算法来识别细胞簇。一旦确定了亚群,通常通过差异表达调用或方差分析(ANOVA)来识别最能区分不同亚群的marker。

ScRNA-Seq数据的差异表达分析(Differential Expression Analysis of ScRNA-Seq Data)
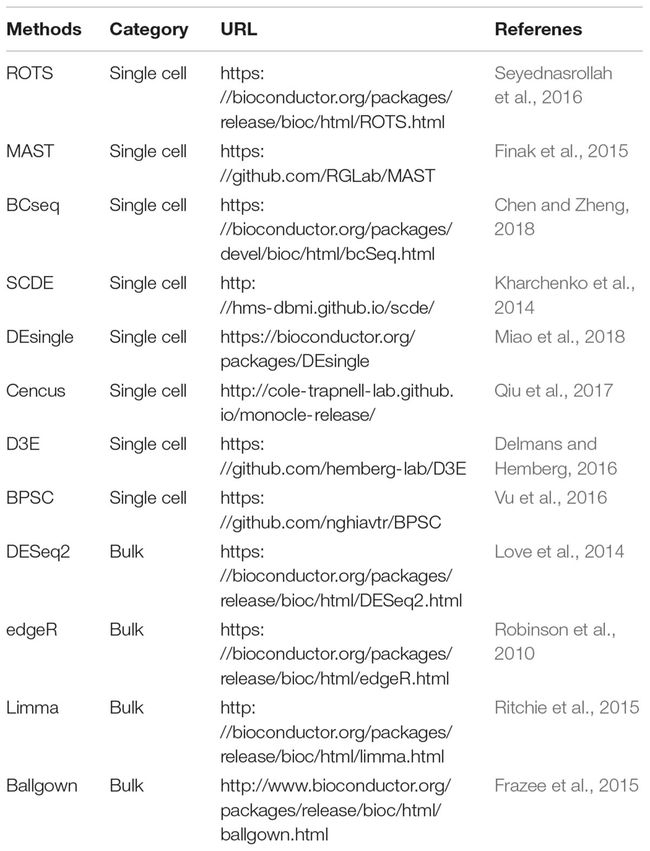
差异表达分析对于发现不同亚群或细胞群间的差异表达基因(DEGs)非常有用。DEGs对于解释两个比较条件之间的生物学差异至关重要。scRNA-seq数据的技术可变性、高噪声(例如,dropout)和大样本规模给差异表达calling带来了挑战。此外,在一个细胞群中可能存在多种细胞状态,导致细胞中基因表达的多样性。最初为 Bulk RNA-seq数据开发的工具已经在许多单细胞研究中用于识别DEGs,但是这些方法对scRNA-seq数据的适用性仍然不清楚。近年来,提出了一些基于scRNA-seq数据进行差异表达基因calling的具体方法,如MAST (Finak et al., 2015)、SCDE (Kharchenko et al., 2014)、DEsingle (Miao et al., 2018)、Census (Qiu et al., 2017)和BCseq (Chen and Zheng,2018)(表4)。MAST是基于线性模型拟合和似然比检验。SCDE是一种贝叶斯方法,使用低强度泊松过程来解释dropouts。DEsingle使用了零膨胀负二项模型来估计损失和实际零。BCseq以数据自适应的方式降低技术噪音。Soneson和Robinson最近评估了36种差异表达方法(包括为scRNA-seq设计的工具和bulk RNA-seq数据),发现这些方法在DEGs的特征和数量上存在显著差异(Soneson and Robinson, 2018)。将开发越来越多的用于scRNA-seq数据差异表达分析的工具,并鼓励用户根据scRNA-seq数据的复杂特性选择专门为scRNA-seq设计的工具来识别DEGs。
细胞谱系和拟时间重构(Cell Lineage and Pseudotime Reconstruction)
许多生物系统中的细胞表现出连续的状态谱,并涉及不同细胞状态之间的转换。通过基于scRNA-seq数据重建细胞轨迹和伪时间,可以对部分细胞内的动态过程进行计算建模。拟时间 (Pseudotime) 是一个系统中沿着一个连续发展过程轨迹的细胞排序,它允许在轨迹的开始、中间和结束状态识别细胞类型(Griffiths et al., 2018)。除了揭示细胞间的基因表达动态外,单细胞轨迹推断还有助于识别触发状态转换的因素。一些工具已经提出了轨迹推理,例如,Monocle(Trapnell et al., 2014),Waterfall(Shin et al.,2015),Wishbone(Setty et al., 2016), TSCAN (Ji and Ji, 2016),Monocle2(Qiu et al ., 2017),Slingshot(Street et al ., 2018),和CellRouter (Lummertz da Rocha et al., 2018)(表5)。由此产生的轨迹拓扑可以是线性的,分叉或一棵树/图结构。Monocle构建了一个最小生成树(MST),用于细胞基于独立成分分析(ICA)搜索最长的主链。Monocle2使用了一种独特的方法,它结合了非监督数据驱动方法和反向图嵌入(RGE),这比Monocle更健壮、更快。Slingshot是一种基于cluster的方法,用于识别具有不同监督级别的多个轨迹。CellRouter利用flow network 来识别细胞状态转换轨迹。最近,Saelens等人(2018)评估了一些单细胞轨迹推断方法(不包括CellRouter),发现Slingshot、TSCAN和Monocle2优于其他方法。

可变剪切和RNA编辑(Alternative Splicing and RNA Editing Analysis of ScRNA-Seq Data)
大多数已发表的单细胞研究主要探讨基因水平上单个细胞之间的转录组差异。在真核生物的基因组中,可变剪切(AS)允许多外显子基因产生不同的亚型,这在很大程度上增加了蛋白质编码和非编码rna的多样性。现有公认的5种基本模式:exon-skipping (cassette exon), mutually exclusive exons, alternative donor site, alternative acceptor site, and intron retention.大量研究表明,AS在哺乳动物中很常见,基于bulk RNA-seq数据,90%以上的人类基因可以发生AS。此外,AS在多种生物学过程中起着关键作用,并且AS异常可能与癌症相关(Sveen et al., 2016)。Bulk RNA-seq 数据揭示的结果只能反映在种群水平上众多细胞的平均模式。由于scRNA-seq数据的高噪音(如dropouts和不均匀的转录覆盖)和低序列覆盖,最初开发的用于Bulk RNA-seq数据的拼接量化方法不适用于scRNA-seq数据。由于表达动态是细胞群体的一个关键方面,因此在单细胞分辨率方面进行研究以了解细胞水平亚型的使用是很有希望的。迄今为止,只有少数的AS检测方法被设计用于scRNA-seq数据,包括SingleSplice (Welch et al., 2016), Census (Qiu et al., 2017), BRIE (Huang and Sanguinetti, 2017), and Expedition (Song et al., 2017) (Table 6).SingleSplice使用统计模型来检测具有显著亚型使用的基因,而不需要估计全长转录本的表达水平。Census以Dirichlet-multinomial分布的线性模式来模拟每个基因的亚型计数。BRIE是一个用于微分亚型量化的贝叶斯层次模型。Expedition包含一套用于识别AS、分配拼接模式和可视化模式更改的算法。

另一方面,RNA编辑是一个重要的转录后处理事件,会导致RNA分子的序列改变(Gott and Emeson, 2000)。类似地,rna编辑主要使用Bulk RNA-seq技术进行研究,但很少在单细胞水平进行研究。目前,scRNA-seq的局限性很大程度上阻碍了rna编辑检测在单个细胞中的应用。因此,随着scRNA-seq技术和单细胞编辑检测算法的发展,探索单细胞间的rna编辑动态将成为可能。值得注意的是,AS和rna编辑主要适用于scRNA-seq protocol生成的数据,这些protocol可以对Smart-seq2和MATQ-seq等测全长转录本文库,而不是3'-end scRNA-seq方法。
等位基因表达探索(Allelic Expression Exploration with ScRNA-Seq Data)
二倍体物种包含两套染色体,分别来自它们的父母。等位基因表达分析可以揭示基因在亲本和母本基因组中的表达是否相等。对于常染色体,亲代和母体的表达通常是平等的,亲代或母体基因组的异常表达可能导致某些疾病(McKean et al., 2016)。迄今为止,基于scRNA-seq数据的全基因组等位基因表达谱检测方法较少。等位基因表达calling需要注意scRNA-seq数据的高dropouts可能会引入许多假阳性。Deng等(2014)在研究小鼠植入前胚胎的等位基因表达谱时,使用了一系列严格的标准来过滤由于scRNA-seq的技术可变性而可能产生的假等位基因。该策略的稳健性在利用小鼠胚胎干细胞分析X染色体失活随发育进程的动力学过程中得到进一步证明(Chen et al., 2016a)。SCALE采用经验贝叶斯方法将基因表达分为沉默状态、单等位基因状态和双等位基因状态(Jiang et al., 2017)。我们认为,单细胞水平的等位基因表达分析可以在很大程度上促进对剂量补偿和相关疾病的潜在机制的理解。值得注意的是,单细胞水平的等位基因表达研究也需要whole-transcript scRNA-seq,主要适用于有父系和母系单核苷酸多态性(SNP)信息的生物体。
基因调控网络重构(Gene Regulatory Network Reconstruction)
基因调控网络推理(Gene regulatory network inference)已经在众多的Bulk RNA-seq研究中得到广泛应用,而scRNA-seq也为此类分析提供了巨大的潜力。对于大量的RNA-seq数据,通常使用加权基因共表达网络分析(WGCNA)等工具从大量样本中构建网络。一个基本的假设是,表达高度相关的基因可以被共同调控。由于这样的分析无法确定监管关系,因此产生的网络通常是无定向的。从理论上讲,scRNA-seq细胞可以作为Bulk RNA-seq的样本,类似的方法也可以应用于scRNA-seq数据构建基因调控网络。
对scRNA-seq数据的网络推断可以揭示有意义的基因相关性,并提供bulk RNA-seq的群体水平数据所不能揭示的生物学上的重要见解。然而,由于scRNA-seq的技术噪声和细胞的不同亚群或状态,网络重构应该引起大家重视。为了减少虚假结果,应该对每个亚群或同一阶段的细胞进行网络推理。最近,Aibar等人(2017)开发了基于scRNA-seq数据重建基因调控网络的SCENIC方法,结果表明,该方法能够较好地预测转录因子与靶基因之间的相互作用。PIDC是另一个利用多元信息论从单细胞数据推断基因调控网络的软件(Chan et al., 2017)。这些网络推理工具有助于从单细胞转录组数据中识别表达调控网络,并为基因间的调控关系提供了批判性的生物学见解。
Conclusion
在过去的10年中,scRNA-seq取得了很大的进步,开发了多种scRNA-seq protocol。scRNA-seq的开发和创新在很大程度上促进了单细胞转录组学的研究,并在细胞表达可变性和动力学方面产生了深刻的发现。此外,随着细胞条形码技术和微流体技术的发展,scRNA-seq的通量也显著提高。同时,最近也提出了可用于固定和冷冻标本的scRNA-seq方法,这将极大地有利于高异质性临床标本的研究。然而,目前可用的scRNA-seq方法仍然存在高退出问题,即弱表达基因可能被遗漏。RNA捕获效率和转录本覆盖率的提高必将降低scRNA-seq的技术噪声。此外,由于目前大多数scRNA-seq方法主要捕获polyA rna,因此,开发能够同时捕获polyA和polyA- rna的protocol(如MATQ-seq)将能够在单细胞分辨率上全面研究蛋白质编码和非编码基因表达动态。
由于scRNA-seq数据的噪声较大,因此采用合适的方法来克服对scRNA-seq数据的分析是至关重要的。质量控制是必要的排除那些低质量的细胞,以避免在数据解释中涉及伪影。此外,批量效应校正(如果需要)、样本归一化和归一化之间也很重要,应该在细胞亚群识别、差异表达调用和其他下游分析之前进行。此外,细胞大小和细胞周期状态等因素可能在某些类型细胞的细胞变异性中发挥重要作用,这些偏差也需要考虑。虽然越来越多的方法被专门设计来解释scRNA-seq数据,但仍需要先进的新方法来有效地处理技术噪音和细胞的表达变异性。具体来说,利用scRNA-seq数据准确分析AS和rna编辑的方法对阐明单个细胞的转录后机制非常有用。综上所述,scRNA-seq数据的生物信息学分析仍然具有挑战性,在数据解释方面需要特别重视,急需更高效的工具。
总的来说,scRNA-seq及其相关的计算方法极大地促进了单细胞转录组学的发展。scRNA-seq技术的不断创新和生物信息学方法的不断进步,将极大地促进生物学和临床研究的发展,为深入了解细胞的基因表达异质性和动态机制提供重要的理论依据。