From: montreal 生信人 5 days ago
CCR5基因编辑婴儿事件已经过去快一年时间了。这一当年闹得沸沸扬扬的争议事件至今仍有很多地方尚未平息。其中一个地方在于CCR5的编辑即使可以成功防止艾滋病,但会否为其携带者增加患其他疾病的风险。今年六月,来自加州大学伯克利分校的Xinzhu Wei和著名计算生物学家Rasmus Nielsen于nature medicine杂志发文,通过群体基因组学分析表明,CCR5的突变(CCR5-∆32)可能会增加过早死亡风险,似乎是“坐实”了这一说法。

然而,风云突变。上月月底,在bioRxiv上,nature medicine这篇文章的原作者就同样主题发布了一篇预印本,题目是“Deviations from Hardy Weinberg Equilibrium at CCR5-Δ32 in Large Sequencing Data Sets?”,文中承认原文存在技术问题(经来自哈佛大学和博德研究所的同事的提醒),还就此事发推致歉。紧接着(实际上是本月二号),bioRxiv上又看到来自哈佛医学院和博德研究所学者的连续发文,指出nature medicine原文存在操作失误,致使结果不可信。
究竟结论如何,还需要几篇文章经过同行评议和时间的检验,而与此同时,大家应该对nature medicine文章的结论十分警惕了。
同行的“纠错”来的迅速,原作者的直面问题也十分值得赞赏。试想如果没有预印本,对于原文潜在错误的纠正也许要经历
a. 向杂志社和作者写信
b. 等待杂志社和作者回应
c. 无回应,goto step a
由于缺乏足够压力和监督,杂志社和原作者有可能一再拖延,最后陷入死循环不了了之。的确,bioRxiv的出现在很大地改变了这一传统的纠错模式。而这种绕过审稿快速po出研究成果供大家分享的方式,不也正是预印本的魅力所在吗?
1. 哈佛医学院David Reich课题组反击:没有证据表明CCR5-Δ32与寿命有关
No statistical evidence for an effect of CCR5-Δ32 on lifespan in the UK Biobank cohort(by-nc-nd 4.0)
A recent study reported that a 32-base-pair deletion in the CCR5 gene (CCR5-Δ32) is deleterious in the homozygous state in humans. Evidence for this came from a survival analysis in the UK Biobank cohort, and from deviations from Hardy-Weinberg equilibrium at a polymorphism tagging the deletion (rs62625034). Here, we carry out a joint analysis of whole-genome genotyping data and whole-exome sequencing data from the UK Biobank, which reveals that technical artifacts are a more plausible cause for deviations from Hardy-Weinberg equilibrium at this polymorphism. Specifically, we find that individuals homozygous for the deletion in the sequencing data are underrepresented in the genotyping data due to an elevated rate of missing data at rs62625034, possibly because the probe for this SNP overlaps with the Δ32 deletion. Another variant which has a higher concordance with the deletion in the sequencing data shows no associations with mortality. A phenome-wide scan for effects of variants tagging this deletion shows an overall inflation of association p-values, but identifies only one trait at p < 5x10-8, and no mediators for an effect on mortality. These analyses show that the original reports of a recessive deleterious effect of CCR5-Δ32 are affected by a technical artifact, and that a closer investigation of the same data provides no positive evidence for an effect on lifespan.
2. 博德研究所Mark J Daly课题组:David Reich说得对
Technical artifact drives apparent deviation from Hardy-Weinberg equilibrium at CCR5-Δ32 and other variants in gnomAD
Following an earlier report suggesting increased mortality due to homozygosity at the CCR5-Δ32 allele, Wei and Nielsen recently suggested a deviation from Hardy-Weinberg Equilibrium (HWE) observed in public variant databases as additional supporting evidence for this hypothesis. Here, we present a re-analysis of the primary data underlying this variant database and identify a pervasive genotyping artifact, especially present at long insertion and deletion polymorphisms. Specifically, very low levels of contamination can affect the variant calling likelihood models, leading to the misidentification of homozygous individuals as heterozygous, and thereby creating an apparent depletion of homozygous calls, which is especially prominent at large insertions and deletions. The deviation from HWE observed at CCR5-Δ32 is a consequence of this specific genotyping error mode rather than a signature of selective pressure at this locus.
3. 原作者发声:存在基因分型错误
Deviations from Hardy Weinberg Equilibrium at CCR5-Δ32 in Large Sequencing Data Sets?(CC-BY-NC-ND 4.0)
We have been made aware by Mark Daly and colleagues that there might be genotyping errors in the gnomeAD database. Also, David Reich and colleagues have pointed out a genotyping error in the UK Biobank that might also affect our results of this paper and certainly has affected our previous paper. For this reason, some of the results in the posted version of this paper may no longer hold up and we ask other researchers to disregard the evidence in this paper pending further inquiry.
4. 牡蛎泛基因组揭示广泛的基因缺失
Massive gene presence/absence variation in the mussel genome as an adaptive strategy: first evidence of a pan-genome in Metazoa
Mussels are ecologically and economically relevant edible marine bivalves, highly invasive and resilient to biotic and abiotic stressors causing recurrent massive mortalities in other species. Here we show that the Mediterranean mussel Mytilus galloprovincialis has a complex pan-genomic architecture, which includes a core set of 45,000 genes shared by all individuals plus a surprisingly high number of dispensable genes (∼15,000). The latter are subject to presence/absence variation (PAV), i.e., they may be entirely missing in a given individual and, when present, they are frequently found as a single copy. The enrichment of dispensable genes in survival functions suggests an adaptive value for PAV, which might be the key to explain the extraordinary capabilities of adaptation and invasiveness of this species. Our study underpins a unique metazoan pan-genome architecture only previously described in prokaryotes and in a few non-metazoan eukaryotes, but that might also characterize other marine invertebrates.
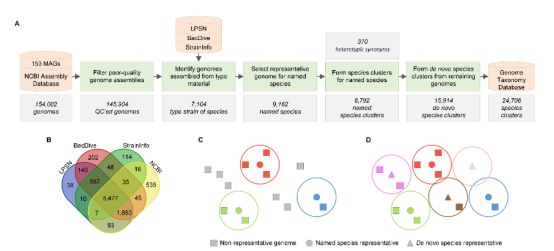
5. 澳洲昆士兰大学Philip Hugenholtz课题组利用95%平均核酸全同率(ANI)全面梳理细菌物种划分
Selection of representative genomes for 24,706 bacterial and archaeal species clusters provide a complete genome-based taxonomy(CC-BY 4.0)
We recently introduced the Genome Taxonomy Database (GTDB), a phylogenetically consistent, genome-based taxonomy providing rank normalized classifications for nearly 150,000 genomes from domain to genus. However, nearly 40% of the genomes used to infer the GTDB reference tree lack a species name, reflecting the large number of genomes in public repositories without complete taxonomic assignments. Here we address this limitation by proposing 24,706 species clusters which encompass all publicly available bacterial and archaeal genomes when using commonly accepted average nucleotide identity (ANI) criteria for circumscribing species. In contrast to previous ANI studies, we selected a single representative genome to serve as the nomenclatural type for circumscribing each species with type strains used where available. We complemented the 8,792 species clusters with validly or effectively published names with 15,914 de novo species clusters in order to assign placeholder names to the growing number of genomes from uncultivated species. This provides the first complete domain to species taxonomic framework which will improve communication of scientific results.
6. 海水鳄鱼湾鳄基因组揭示咸水适应
A high-quality reference genome assembly of the saltwater crocodile, Crocodylus porosus, reveals patterns of selection in Crocodylidae(CC-BY-NC-ND 4.0)
Crocodilians are an economically, culturally, and biologically important group. To improve researchers’ ability to study genome structure, evolution, and gene regulation in the clade, we generated a high-quality de novo genome assembly of the saltwater crocodile, Crocodylus porosus, from Illumina short read data from genomic libraries and in vitro proximity-ligation libraries. The assembled genome is 2,123.5 Mb, with N50 scaffold size of 17.7 Mb and N90 scaffold size of 3.8 Mb. We then annotated this new assembly, increasing the number of annotated genes by 74%. In total, 96% of 23,242 annotated genes were associated with a functional protein domain. Furthermore, multiple non-coding functional regions and mappable genetic markers were identified. Upon analysis and overlapping the results of branch length estimation and site selection tests for detecting potential selection, we found 16 putative genes under positive selection in crocodilians, ten in C. porosus and six in A. mississippiensis. The annotated C. porosus genome will serve as an important platform for osmoregulatory, physiological and sex determination studies, as well as an important reference in investigating the phylogenetic relationships of crocodilians, birds, and other tetrapods.

7. 斯坦福大学学者:癌症基因组范围内的连锁导致选择的降低
Most cancers carry a substantial deleterious load due to Hill-Robertson interference(CC-BY-NC-ND 4.0)
Results We find that appreciable negative selection (dN/dS ~ 0.4) is present in tumors with a low mutational burden, while the remaining cancers (96%) exhibit dN/dS ratios approaching 1, suggesting that the majority of tumors do not remove deleterious passengers. A parallel pattern is seen in drivers, where positive selection attenuates as the mutational burden of cancers increases. Both trends persist across tumor-types, are not exclusive to essential or housekeeping genes, and are present in clonal and subclonal mutations. Two additional orthogonal lines of evidence support the weak efficacy model: passengers are less damaging in low mutational burden cancers, and patterns of attenuated selection also emerge in Copy Number Alterations. Finally, we find that an evolutionary model incorporating Hill-Robertson interference can reproduce both patterns of attenuated selection in drivers and passengers if the average fitness cost of passengers is 1.0% and the average fitness benefit of drivers is 19%.
8. 范德堡大学(Vanderbilt University)学者基于单细胞RNA测序的肺纤维化研究
Single-cell RNA-sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis(CC-BY-NC-ND 4.0)
One Sentence Summary Single-cell RNA-sequencing provides new insights into pathologic epithelial and mesenchymal remodeling in the human lung.
9. 芝加哥大学Savaş Tay等人:无氧条件下对于稀有肠道微生物的高通量分拣
High-throughput isolation and sorting of gut microbes reduce biases of traditional cultivation strategies
Traditional cultivation approaches in microbiology are labor-intensive, low-throughput, and often yield biased sampling of taxa due to ecological and evolutionary factors. New strategies are needed to enable ample representation of rare taxa and slow-growers that are outcompeted by fast-growing organisms. We developed a microfluidic platform that anaerobically isolates and cultivates microbial cells in millions of picoliter droplets and automatically sorts droplets based on colony density. We applied our strategy to mouse and human gut microbiomes and used 16S ribosomal RNA gene amplicons to characterize taxonomic composition of cells grown using different media. We found up to 4-fold increase in richness and larger representation of rare taxa among cells grown in droplets compared to conventional culture plates. Automated sorting of droplets for slow-growing colonies further enhanced the relative abundance of rare populations. Our method improves the cultivation and analysis of diverse microbiomes to gain deeper insights into microbial functioning and lifestyles.
10. 澳洲新南威尔士大学(university of new south wales)Sri Parameswaran实验室开发Nanopore 信号的高效分析软件f5c
GPU Accelerated Adaptive Banded Event Alignment for Rapid Comparative Nanopore Signal Analysis(CC-BY-NC 4.0)
Nanopore sequencing has the potential to revolutionise genomics by realising portable, real-time sequencing applications, including point-of-care diagnostics and in-the-field genotyping. Achieving these applications requires efficient bioinformatic algorithms for the analysis of raw nanopore signal data. For instance, comparing raw nanopore signals to a biological reference sequence is a computationally complex task despite leveraging a dynamic programming algorithm for Adaptive Banded Event Alignment (ABEA)—a commonly used approach to polish sequencing data and identify non-standard nucleotides, such as measuring DNA methylation. Here, we parallelise and optimise an implementation of the ABEA algorithm (termed f5c) to efficiently run on heterogeneous CPU-GPU architectures. By optimising memory, compute and load balancing between CPU and GPU, we demonstrate how f5c can perform ~3-5× faster than the original implementation of ABEA in the Nanopolish software package. We also show that f5c enables DNA methylation detection on-the-fly using an embedded System on Chip (SoC) equipped with GPUs. Our work not only demonstrates that complex genomics analyses can be performed on lightweight computing systems, but also benefits High-Performance Computing (HPC). The associated source code for f5c along with GPU optimised ABEA is available at https://github.com/hasindu2008/f5c.

原载于生信人公众号
