刘小泽写于19.10.26-27,更新于2020-06-23,最近会继续整理这本资料
为何取名叫“交响乐”?因为单细胞分析就像一个大乐团,需要各个流程的协同配合分析单细胞数据,常见的一个名称就是
SingleCellExperiment或者sce,那么这次就来认识一下这个基础知识点
前言
我们首先要对单细胞分析的流程有一个大概的认识:
上半场分析:
这个差不多属于固定的流程了
首先是数据导入
紧接着进行质量控制和归一化,这个步骤包含了矫正实验因素以及其他影响因素,目的是从raw count得到clean count
-
有了clean count,就要挑取导致表达量差异的基因们(用于后面的降维)
因为降维的过程其实就是去繁存简,每个基因的变化都对整体有影响,对细胞来讲都是一个变化维度。但这么多维度我们看不了也处理不了,需要尽可能保证真实差异的前提下减少维度的数量,因此需要挑出那些更能代表整体差异的基因
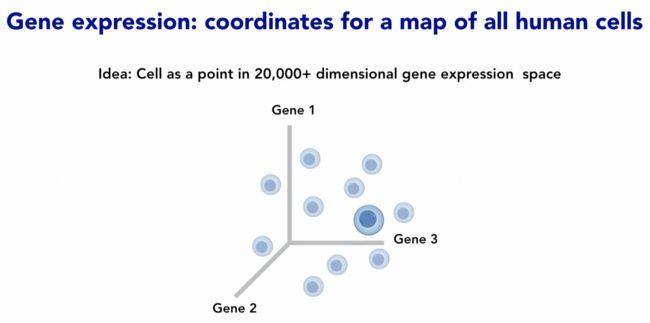
降维
下半场分析:
这个就可以分出很多分支,例如
- 分群(clustering):意在探索如何把scRNA数据集给拆分掉,变成一小块一小块的数据,这每一小块基因变化都是相似的
- 差异分析(differential expression):不同组的细胞之间表达量差异是如何产生的
- 数据集的整合(Integrating datasets):scRNA数据集越来越多,数据集之间的比较也日益显著
- 处理大型数据(large scale data):这部分仅仅依靠内存存储的数据是不够的,还有更好的办法
- 还有:轨迹推断、细胞周期推断等等特定需求
知道了大体的流程,那么就先看看必须了解的数据结构
这是单细胞分析中的非常常用的S4对象,里面包罗万象,但依然有据可循。那么它是如何组织的?存储了什么内容?这就是我们这次要探索的任务。内容来自:https://osca.bioconductor.org/data-infrastructure.html
首先要上一张图
这张图会不断反复查看
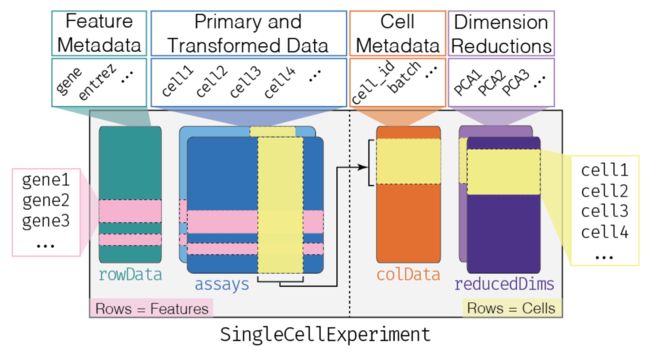
图中最核心的部分,是蓝色的data部分;另外还有绿色的基因注释信息feature metadata、橙色的细胞注释信息cell metadata。除了这三大件,还会包含一些下游分析结果,比如PCA、tSNE降维结果就会保存在紫色的Dimension Reductions
这个SingleCellExperiment对象来自SingleCellExperimentR包,现在Bioconductor上的70多个单细胞相关的R包都使用了这个对象,可以说是单细胞领域的通用货币,包含了: gene-by-cell expression data、 per-cell metadata 、 per-gene annotation
开始发挥想象力了哈!
如果把这个SingleCellExperiment对象比作一艘货船,上面就会装载许多集装箱slots(理解为对象结构) 。每个箱子slot都是独立的,箱子里面包含的东西不一样,比如有的装食物,有的装砖头,各有各的特征。就sce来说,有的接口必须是数值型矩阵结构,有的就需要数据框结构。
核心部分-assays
创建一个sce对象很容易,只需要一个assays就可以,这是一个列表,其中包含了许多表达数据,例如原始数据count或者其他标准化处理过的数据,行是基因,列是样本
可以构建一个10个基因,3个细胞的矩阵【一定要是矩阵】
counts_matrix <- data.frame(cell_1 = rpois(10, 10),
cell_2 = rpois(10, 10),
cell_3 = rpois(10, 30))
rownames(counts_matrix) <- paste0("gene_", 1:10)
counts_matrix <- as.matrix(counts_matrix)
有了这个,就可以用一个list构建出SingleCellExperiment对象。当然,这个list中可以包括任意个矩阵
# BiocManager::install('SingleCellExperiment')
sce <- SingleCellExperiment(assays = list(counts = counts_matrix))
>sce
## class: SingleCellExperiment
## dim: 10 3
## metadata(0):
## assays(1): counts
## rownames(10): gene_1 gene_2 ... gene_9 gene_10
## rowData names(0):
## colnames(3): cell_1 cell_2 cell_3
## colData names(0):
## reducedDimNames(0):
## spikeNames(0):
## altExpNames(0):
为了得到这个对象中的矩阵,可以用两种方式获得:
-
assay(sce, "counts"):这个方法是最通用的办法,而且其中的counts可以换成其他的名称,只要是出现在之前的list中都可以 -
counts(sce):它实现的东西和上面一样,只不过这个方法只适合提取counts这个名称的矩阵
counts(sce)
# 或者assay(sce, "counts")
## cell_1 cell_2 cell_3
## gene_1 7 9 35
## gene_2 7 6 38
## gene_3 10 14 32
## gene_4 7 9 32
## gene_5 19 19 48
## gene_6 8 7 26
## gene_7 10 10 28
## gene_8 4 10 26
## gene_9 10 9 37
## gene_10 6 16 26
assays还能扩展
既然有了核心,那么就可以根据它进行多向拓展,这也是它强大的一个原因。
使用标准函数扩展
之前assays中只有原始表达矩阵,其实还能根据它扩展到归一化矩阵,例如使用一些R包的函数对包装的矩阵进行操作:
sce <- scran::computeSumFactors(sce)
sce <- scater::normalize(sce)
> sce
## class: SingleCellExperiment
## dim: 10 3
## metadata(1): log.exprs.offset
## assays(2): counts logcounts
## rownames(10): gene_1 gene_2 ... gene_9 gene_10
## rowData names(0):
## colnames(3): cell_1 cell_2 cell_3
## colData names(0):
## reducedDimNames(0):
## spikeNames(0):
## altExpNames(0):
这样,assays 就从一个存储原始矩阵的counts ,又扩增了归一化矩阵的logcounts 。同理,这个logcounts也是能有两种提取方法:
logcounts(sce)
# assay(sce, "logcounts")
## cell_1 cell_2 cell_3
## gene_1 3.90 3.95 4.30
## gene_2 3.90 3.41 4.41
## gene_3 4.38 4.55 4.18
## gene_4 3.90 3.95 4.18
## gene_5 5.28 4.98 4.73
## gene_6 4.08 3.61 3.89
## gene_7 4.38 4.09 3.99
## gene_8 3.16 4.09 3.89
## gene_9 4.38 3.95 4.37
## gene_10 3.69 4.74 3.89
通过对比这个logcounts和counts数据,就能发现为什么要做normalization这一步:原始矩阵中1、2表达量差不多,但和3差别很大,很有可能是细胞3本身测序深度就比较高,因此得到的reads数也多;进行归一化以后,应该就去除了样本文库差异,结果看到1、2、3之间也变得可比了
到目前为止,我们总共得到了:
assays(sce)
## List of length 2
## names(2): counts logcounts
自定义扩展assays
这里的自定义指的是,我们不使用某个R包的某个函数,而是根据自己的想法,去根据原始矩阵得到一个新的矩阵
# 例如自己创建一个新的counts_100矩阵,然后依旧是通过这个名称进行访问
counts_100 <- assay(sce, "counts") + 100
assay(sce, "counts_100") <- counts_100
看一下结果【注意:新增用的是assay单数,查看结果用的是assays复数】
assays(sce)
## List of length 3
## names(3): counts logcounts counts_100
小结
再回到第一张图,看看assays那里,是不是画了深蓝和浅蓝?这也是为了更好地表达:assays可以包含多个矩阵;构建sce对象可以一次一次加入新的矩阵,也可以用列表的形式,一次加入多个矩阵
列的注释信息:colData
之前有了”核心“——表达矩阵信息,那么其次重要的就是添加注释信息,这部分来介绍列的注释,针对的就是实验样本、细胞。这部分信息将会保存在colData中,它的主体是样本,于是将行名设定为样本,列名设为注释信息(如:批次、作者等等),对应上面图中的橙色部分。
接下来先设置一个细胞批次注释信息:
cell_metadata <- data.frame(batch = c(1, 1, 2))
rownames(cell_metadata) <- paste0("cell_", 1:3)
有了注释信息,怎么把它加入sce对象呢?
两种方法:一种是直接构建,一种是后续添加
-
直接构建:
sce <- SingleCellExperiment(assays = list(counts = counts_matrix), colData = cell_metadata) -
后续添加
colData(sce) <- DataFrame(cell_metadata)
然后看看sce对象添加了什么:
可以看到colData增加了之前设置的batch信息
sce
## class: SingleCellExperiment
## dim: 10 3
## metadata(0):
## assays(1): counts
## rownames(10): gene_1 gene_2 ... gene_9 gene_10
## rowData names(0):
## colnames(3): cell_1 cell_2 cell_3
## colData names(1): batch
## reducedDimNames(0):
## spikeNames(0):
## altExpNames(0):
加入了sce对象以后,怎么获取它呢?
colData(sce)
## DataFrame with 3 rows and 1 column
## batch
##
## cell_1 1
## cell_2 1
## cell_3 2
或者直接看结果信息:
sce$batch
## [1] 1 1 2
这个注释信息只能自己手动添加吗?
答案是no!有一些包可以自己计算,并且帮你添加进去。例如scater包的calculateQCMetrics()就会帮你计算几十项细胞的质量信息,结果依然是使用colData调用注释结果信息
sce <- scater::addPerCellQC(sce)
colData(sce)[, 1:5]
## DataFrame with 3 rows and 5 columns
## batch sum detected percent_top_50 percent_top_100
##
## cell_1 1 80 10 100 100
## cell_2 1 88 10 100 100
## cell_3 2 309 10 100 100
还能任意添加
sce$more_stuff <- runif(ncol(sce))
colnames(colData(sce))
sce$more_stuff
## 0.04094262 0.41983904 0.51959352
既然colData可以包含这么多的注释信息,那么怎么从中选取一部分呢?
colData的一个常用操作就是取子集,看下面操作:
例如想从colData中选择批次信息,和数据框取子集是类似的
sce$batch
# 或者colData(sce)$batch
## [1] 1 1 2
然后如果再想取批次中属于第一个批次的信息,就可以继续按照数据框的思路取子集:
sce[, sce$batch == 1]
## class: SingleCellExperiment
## dim: 10 2
## metadata(0):
## assays(1): counts
## rownames(10): gene_1 gene_2 ... gene_9 gene_10
## rowData names(0):
## colnames(2): cell_1 cell_2
## colData names(9): batch sum ... total more_stuff
## reducedDimNames(0):
## altExpNames(0):
这样看的colnames中就只剩两个细胞了
行的注释信息:rowData/rowRanges
既然样本有注释信息,那么同样的,基因也有自己的注释,它就存放在rowData或者rowRanges中,这两个的区别就是:
-
rowData:是一个数据框的结构,它就存储了核心assays矩阵的基因相关信息它返回的结果就是这样:
# 一开始rowData(sce)是空的,可以添加 sce <- scater::addPerFeatureQC(sce) rowData(sce) ## DataFrame with 10 rows and 2 columns ## mean detected #### gene_1 16.0000 100 ## gene_2 14.3333 100 ## gene_3 16.0000 100 ## gene_4 18.6667 100 ## gene_5 15.3333 100 ## gene_6 16.6667 100 ## gene_7 17.6667 100 ## gene_8 13.0000 100 ## gene_9 16.6667 100 ## gene_10 14.6667 100
-
rowRanges:也是基因相关,但是它是GRange对象,存储了基因坐标信息,例如染色体信息、起始终点坐标它返回的结果一开始是空的:
rowRanges(sce) ## GRangesList object of length 10: ## $gene_1 ## GRanges object with 0 ranges and 0 metadata columns: ## seqnames ranges strand #### ------- ## seqinfo: no sequences ## ## $gene_2 ## GRanges object with 0 ranges and 0 metadata columns: ## seqnames ranges strand ## ## ------- ## seqinfo: no sequences ## ## $gene_3 ## GRanges object with 0 ranges and 0 metadata columns: ## seqnames ranges strand ## ## ------- ## seqinfo: no sequences ## ## ... ## <7 more elements>
怎么按行取子集?
同样类似于数据框,可以按位置、名称取子集:
sce[c("gene_1", "gene_4"), ]
# 或者 sce[c(1, 4), ]
## class: SingleCellExperiment
## dim: 2 3
## metadata(0):
## assays(1): counts
## rownames(2): gene_1 gene_4
## rowData names(7): is_feature_control mean_counts ... total_counts
## log10_total_counts
## colnames(3): cell_1 cell_2 cell_3
## colData names(10): batch is_cell_control ...
## pct_counts_in_top_200_features pct_counts_in_top_500_features
## reducedDimNames(0):
## spikeNames(0):
## altExpNames(0):
看到rownames结果就剩2个基因
除了以上3大块,还有一些重要的”集装箱“需要介绍,这些前面用
附标识
附1:对样本进行归一化:sizeFactors
这里面装了根据样本文库计算的文库大小因子,是一个数值型向量,用于后面的归一化
sce <- scran::computeSumFactors(sce)
sizeFactors(sce)
## [1] 0.5031447 0.5534591 1.9433962
# 或者手动添加
sizeFactors(sce) <- scater::librarySizeFactors(sce)
sizeFactors(sce)
## cell_1 cell_2 cell_3
## 0.5031447 0.5534591 1.9433962
前面提到的: assays (primary data), colData (sample metadata), rowData/rowRanges (feature metadata), and sizeFactors 。其实这其中前三个都来自于SummarizedExperiment这个对象。基于这个对象,还建立了一些新的对象接口,例如下面的:
附2:降维结果:reducedDims
存储了原始矩阵的降维结果,可以通过PCA、tSNE、UMAP等得到,它是一个数值型矩阵的list,行名是原来矩阵的列名(就是细胞、样本),它的列就是各种维度信息
它和assays一样,也可以包含许多降维的结果,例如用scater包计算PCA:
sce <- scater::logNormCounts(sce)
sce <- scater::runPCA(sce)
# 这个算法是利用了sce对象的归一化结果logcounts(sce)
reducedDim(sce, "PCA")
# PC1 PC2
# cell_1 -0.6897959 -0.4080860
# cell_2 0.8493039 -0.2145128
# cell_3 -0.1595080 0.6225988
# attr(,"percentVar")
# [1] 67.07301 32.92699
除了PCA,tSNE的结果也是存储在这里:
sce <- scater::runTSNE(sce, perplexity = 0.1)
reducedDim(sce, "TSNE")
## [,1] [,2]
## cell_1 35.64851 5694.164
## cell_2 -4951.13619 -2815.978
## cell_3 4915.48768 -2878.186
看下全部的结果都包含什么:
reducedDims(sce)
## List of length 2
## names(2): PCA TSNE
调取方法也十分类似:assay,数据矩阵存储在assays,而调用是assay;这里的降维结果存储在reducedDims,调用是reducedDim
这个降维信息只能自己手动添加吗?
答案也是no!和前面的counts_100加到assays的思路一样,我们也可以自己计算,而不用现成的函数,最后加到reducedDims这个接口。
例如,进行UMAP降维,虽然可以用scater::runUMAP(),但依然可以自己处理。
比如这里使用uwot包,不过这个包只能计算,不能添加到sce对象,需要手动添加:
u <- uwot::umap(t(logcounts(sce)), n_neighbors = 2)
reducedDim(sce, "UMAP_uwot") <- u
## [,1] [,2]
## cell_1 -0.3952368 -0.03182602
## cell_2 0.2347576 0.60432243
## cell_3 0.1604792 -0.57249641
## attr(,"scaled:center")
## [1] 7.596012 19.251450
最后再来看一下:
reducedDims(sce)
## List of length 3
## names(3): PCA TSNE UMAP_uwot
附3:metadata 接口
虽然前面介绍了许多接口,但还是有很多DIY的,不能直接导入它们,不过我们仍然需要这些信息,于是medata接口诞生。它可以存储任意类型的数据,只要给它一个名字。
例如,我们有几个感兴趣的基因(比如是高变化基因),现在想要把它保存在sce中,方便以后使用:
my_genes <- c("gene_1", "gene_5")
metadata(sce) <- list(favorite_genes = my_genes)
metadata(sce)
## $favorite_genes
## [1] "gene_1" "gene_5"s
既然是一个列表,就意味着支持扩展:
your_genes <- c("gene_4", "gene_8")
metadata(sce)$your_genes <- your_genes
metadata(sce)
## $favorite_genes
## [1] "gene_1" "gene_5"
##
## $your_genes
## [1] "gene_4" "gene_8"
附4:spike-in信息
还是可以继续使用isSpike来添加,虽然会显示'isSpike' is deprecated:
isSpike(sce, "ERCC") <- grepl("^ERCC-", rownames(sce))
# 结果就会在sce中添加:
## spikeNames(1): ERCC
spikeNames(sce)
## [1] "ERCC"
table(isSpike(sce, "ERCC"))
# 就能看存在多少Spike-in
现在推出了:alternative Experiments
spike_counts <- cbind(cell_1 = rpois(5, 10),
cell_2 = rpois(5, 10),
cell_3 = rpois(5, 30))
rownames(spike_counts) <- paste0("spike_", 1:5)
spike_se <- SummarizedExperiment(list(counts=spike_counts))
spike_se
## class: SummarizedExperiment
## dim: 5 3
## metadata(0):
## assays(1): counts
## rownames(5): spike_1 spike_2 spike_3 spike_4 spike_5
## rowData names(0):
## colnames(3): cell_1 cell_2 cell_3
## colData names(0):
然后通过altExp()加入进来,它的操作和assays或reducedDims()类似:
altExp(sce, "spike") <- spike_se
altExps(sce)
## List of length 1
## names(1): spike
# 提取数据也是类似的
sub <- sce[,1:2] # retain only two samples.
altExp(sub, "spike")
## class: SummarizedExperiment
## dim: 5 2
## metadata(0):
## assays(1): counts
## rownames(5): spike_1 spike_2 spike_3 spike_4 spike_5
## rowData names(0):
## colnames(2): cell_1 cell_2
## colData names(0):
附5:对列添加label
使用colLabels() ,尤其在非监督聚类过程中对细胞添加label,进行分组
colLabels(sce) <- LETTERS[1:3]
colLabels(sce)
## [1] "A" "B" "C"
例如scran::findMarkers就是通过colLabels()来提取细胞信息的
小结
SingleCellExperiment对象兼容性很好,可以用于多个scRNA的R包的输入数据
中间分析的每一步都会向这个对象的assays, colData, reducedDims 等模块添加信息
这个对象方便了未来数据传输与协作,这本书的后续也会基于这个对象进行探讨
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
