
https://doi.org/10.1093/bioinformatics/btaa976
Abstract
Motivation
Single-cell RNA-sequencing (scRNA-seq) offers the opportunity to dissect heterogeneous cellular compositions and interrogate the cell-type-specific gene expression patterns across diverse conditions. However, batch effects such as laboratory conditions and individual-variability hinder their usage in cross-condition designs.
Results
Here, we present a single-cell Generative Adversarial Network (scGAN) to simultaneously acquire patterns from raw data while minimizing the confounding effect driven by technical artifacts or other factors inherent to the data. Specifically, scGAN models the data likelihood of the raw scRNA-seq counts by projecting each cell onto a latent embedding. Meanwhile, scGAN attempts to minimize the correlation between the latent embeddings and the batch labels across all cells. We demonstrate scGAN on three public scRNA-seq datasets and show that our method confers superior performance over the state-of-the-art methods in forming clusters of known cell types and identifying known psychiatric genes that are associated with major depressive disorder.

key: why?单细胞数据存在批次效应(实验室条件和个体差异)
how? VAE+GAN
which is generator?
which is discriminator?
why can it remove batch effect?
编码器网络还需要最小化每个单元样本的嵌入和混淆批次变量之间的相关性。 另一方面,鉴别器通过使用编码器生成的嵌入作为输入来学习预测批次变量


key: why? 寻找阿兹海默症的候选药物
how?
首先,我们收集了561个据报道是AD风险基因的基因,并对这些基因进行了功能富集分析。 然后,通过基于人类相互作用组定量5595种分子药物与AD之间的接近度,我们筛选出了1092种与疾病最接近的药物。 我们进一步对这些候选药物进行了反向基因集富集分析,这使我们能够估计扰动对基因表达的影响,并确定24种潜在的AD治疗候选药物。

Abstract
Drug repurposing involves the identification of new applications for existing drugs at a lower cost and in a shorter time. There are different computational drug-repurposing strategies and some of these approaches have been applied to the coronavirus disease 2019 (COVID-19) pandemic. Computational drug-repositioning approaches applied to COVID-19 can be broadly categorized into (i) network-based models, (ii) structure-based approaches and (iii) artificial intelligence (AI) approaches. Network-based approaches are divided into two categories: network-based clustering approaches and network-based propagation approaches. Both of them allowed to annotate some important patterns, to identify proteins that are functionally associated with COVID-19 and to discover novel drug–disease or drug–target relationships useful for new therapies. Structure-based approaches allowed to identify small chemical compounds able to bind macromolecular targets to evaluate how a chemical compound can interact with the biological counterpart, trying to find new applications for existing drugs. AI-based networks appear, at the moment, less relevant since they need more data for their application.
key: 1)基于网络(聚类和传播) 2)基于结构 3)基于AI

基于网络的聚类:modules
基于网络的传播:提示关键过程是病毒刺突蛋白与人血管紧张素转化酶2(ACE2)和跨膜丝氨酸蛋白酶2(TMPRSS2)的相互作用:刺突蛋白的受体结合结构域与人ACE2的肽酶结构域结合。Mpro介导病毒的复制和转录
计算了药物靶标与HCoV相关蛋白之间的网络邻近度,以筛选人蛋白相互作用组模型下HCoV的候选可重复使用药物

Abstract
A newly described coronavirus named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is the causative agent of coronavirus disease 2019 (COVID-19), has infected over 2.3 million people, led to the death of more than 160,000 individuals and caused worldwide social and economic disruption. There are no antiviral drugs with proven clinical efficacy for the treatment of COVID-19, nor are there any vaccines that prevent infection with SARS-CoV-2, and efforts to develop drugs and vaccines are hampered by the limited knowledge of the molecular details of how SARS-CoV-2 infects cells. Here we cloned, tagged and expressed 26 of the 29 SARS-CoV-2 proteins in human cells and identified the human proteins that physically associated with each of the SARS-CoV-2 proteins using affinity-purification mass spectrometry, identifying 332 high-confidence protein–protein interactions between SARS-CoV-2 and human proteins. Among these, we identify 66 druggable human proteins or host factors targeted by 69 compounds (of which, 29 drugs are approved by the US Food and Drug Administration, 12 are in clinical trials and 28 are preclinical compounds). We screened a subset of these in multiple viral assays and found two sets of pharmacological agents that displayed antiviral activity: inhibitors of mRNA translation and predicted regulators of the sigma-1 and sigma-2 receptors. Further studies of these host-factor-targeting agents, including their combination with drugs that directly target viral enzymes, could lead to a therapeutic regimen to treat COVID-19.
https://www.nature.com/articles/s41586-020-2286-9
key: 26 332 66 69 2
如何实验?
克隆表达了新冠病毒29种蛋白当中的26个蛋白,亲和纯化质谱法鉴定出与新冠病毒蛋白有相互作用的人类蛋白,找到332个高置信度的蛋白-蛋白相互作用关系。找到其中是药物靶点的66个蛋白,共有69种化合物。我们在多种病毒分析中筛选了其中的一个子集,发现了两组具有抗病毒活性的药理剂:mRNA翻译抑制剂和sigma-1和sigma-2受体的调节剂。


Abstract
The recent epidemic outbreak of a novel human coronavirus called SARS-CoV-2 causing the respiratory tract disease COVID-19 has reached worldwide resonance and a global effort is being undertaken to characterize the molecular features and evolutionary origins of this virus. In this paper, we set out to shed light on the SARS-CoV-2/host receptor recognition, a crucial factor for successful virus infection. Based on the current knowledge of the interactome between SARS-CoV-2 and host cell proteins, we performed Master Regulator Analysis to detect which parts of the human interactome are most affected by the infection. We detected, amongst others, affected apoptotic and mitochondrial mechanisms, and a downregulation of the ACE2 protein receptor, notions that can be used to develop specific therapies against this new virus.
key: 125 proteins (31 viral proteins and 94 human host proteins) and 200 unique interactions.
https://www.mdpi.com/2077-0383/9/4/982/htm#B28-jcm-09-00982
key: 病毒感染过程中,哪些蛋白相互作用更容易被感染影响
how? 把病毒-人类蛋白相互关系,映射到基因调控网络,利用master regulator analysis分析关键的调控因子


Abstract
Coronavirus Disease-2019 (COVID-19) is an infectious disease caused by the SARS-CoV-2 virus. Various studies exist about the molecular mechanisms of viral infection. However, such information is spread across many publications and it is very time-consuming to integrate, and exploit. We develop CoVex, an interactive online platform for SARS-CoV-2 host interactome exploration and drug (target) identification. CoVex integrates virus-human protein interactions, human protein-protein interactions, and drug-target interactions. It allows visual exploration of the virus-host interactome and implements systems medicine algorithms for network-based prediction of drug candidates. Thus, CoVex is a resource to understand molecular mechanisms of pathogenicity and to prioritize candidate therapeutics. We investigate recent hypotheses on a systems biology level to explore mechanistic virus life cycle drivers, and to extract drug repurposing candidates. CoVex renders COVID-19 drug research systems-medicine-ready by giving the scientific community direct access to network medicine algorithms. It is available at https://exbio.wzw.tum.de/covex/.
https://www.nature.com/articles/s41467-020-17189-2
key: 它可以对病毒-宿主相互作用组进行可视化探索,并实现系统医学算法,用于基于网络的候选药物预测

AI方法:

https://www.sciencedirect.com/science/article/pii/S2319417020300494
Abstract
Background
The ongoing COVID-19 pandemic has caused more than 193,825 deaths during the past few months. A quick-to-be-identified cure for the disease will be a therapeutic medicine that has prior use experiences in patients in order to resolve the current pandemic situation before it could become worsening. Artificial intelligence (AI) technology is hereby applied to identify the marketed drugs with potential for treating COVID-19.
Methods
An AI platform was established to identify potential old drugs with anti-coronavirus activities by using two different learning databases; one consisted of the compounds reported or proven active against SARS-CoV, SARS-CoV-2, human immunodeficiency virus, influenza virus, and the other one containing the known 3C-like protease inhibitors. All AI predicted drugs were then tested for activities against a feline coronavirus in in vitro cell-based assay. These assay results were feedbacks to the AI system for relearning and thus to generate a modified AI model to search for old drugs again.
Results
After a few runs of AI learning and prediction processes, the AI system identified 80 marketed drugs with potential. Among them, 8 drugs (bedaquiline, brequinar, celecoxib, clofazimine, conivaptan, gemcitabine, tolcapone, and vismodegib) showed in vitro activities against the proliferation of a feline infectious peritonitis (FIP) virus in Fcwf-4 cells. In addition, 5 other drugs (boceprevir, chloroquine, homoharringtonine, tilorone, and salinomycin) were also found active during the exercises of AI approaches.
Conclusion
Having taken advantages of AI, we identified old drugs with activities against FIP coronavirus. Further studies are underway to demonstrate their activities against SARS-CoV-2 in vitro and in vivo at clinically achievable concentrations and doses. With prior use experiences in patients, these old drugs if proven active against SARS-CoV-2 can readily be applied for fighting COVID-19 pandemic.
key:
建立了一个AI平台,通过使用两个不同的学习数据库来识别具有抗冠状病毒活性的潜在旧药物; 一种由已报道或证明对SARS-CoV,SARS-CoV-2,人免疫缺陷病毒,流感病毒具有活性的化合物组成,另一种包含已知的3C样蛋白酶抑制剂。 然后在基于细胞的体外试验中测试所有AI预测的药物对猫冠状病毒的活性。 这些测定结果被反馈到AI系统进行再学习,从而生成修改后的AI模型以再次搜索旧药。
why? 猫冠状病毒是一种α-冠状病毒,是在家猫和野猫中引起肠炎的病毒。 大约5–15%的感染猫患上猫传染性腹膜炎(FIP),对猫是致命的[2]。 猫中FIP病毒的感染表现出与严重急性呼吸系统综合症(SARS)感染类似的特征,例如人的肺部病变[3]。 据证明,核苷类似物GS-441524和3C样蛋白酶抑制剂GC376均在体外表现出对FIP病毒的抗病毒活性,可有效治疗猫的FIP。


Abstract
The infection of a novel coronavirus found in Wuhan of China (SARS-CoV-2) is rapidly spreading, and the incidence rate is increasing worldwide. Due to the lack of effective treatment options for SARS-CoV-2, various strategies are being tested in China, including drug repurposing. In this study, we used our pre-trained deep learning-based drug-target interaction model called Molecule Transformer-Drug Target Interaction (MT-DTI) to identify commercially available drugs that could act on viral proteins of SARS-CoV-2. The result showed that atazanavir, an antiretroviral medication used to treat and prevent the human immunodeficiency virus (HIV), is the best chemical compound, showing an inhibitory potency with Kd of 94.94 nM against the SARS-CoV-2 3C-like proteinase, followed by remdesivir (113.13 nM), efavirenz (199.17 nM), ritonavir (204.05 nM), and dolutegravir (336.91 nM). Interestingly, lopinavir, ritonavir, and darunavir are all designed to target viral proteinases. However, in our prediction, they may also bind to the replication complex components of SARS-CoV-2 with an inhibitory potency with Kd < 1000 nM. In addition, we also found that several antiviral agents, such as Kaletra (lopinavir/ritonavir), could be used for the treatment of SARS-CoV-2. Overall, we suggest that the list of antiviral drugs identified by the MT-DTI model should be considered, when establishing effective treatment strategies for SARS-CoV-2.
key: 深度学习预测药物-靶点相互作用。药物表示为SMILE(化合物的一维结构),靶点表示为氨基酸序列,序列模型。Transformer.

Summary
We performed RNA-seq and high-resolution mass spectrometry on 128 blood samples from COVID-19-positive and COVID-19-negative patients with diverse disease severities and outcomes. Quantified transcripts, proteins, metabolites, and lipids were associated with clinical outcomes in a curated relational database, uniquely enabling systems analysis and cross-ome correlations to molecules and patient prognoses. We mapped 219 molecular features with high significance to COVID-19 status and severity, many of which were involved in complement activation, dysregulated lipid transport, and neutrophil activation. We identified sets of covarying molecules, e.g., protein gelsolin and metabolite citrate or plasmalogens and apolipoproteins, offering pathophysiological insights and therapeutic suggestions. The observed dysregulation of platelet function, blood coagulation, acute phase response, and endotheliopathy further illuminated the unique COVID-19 phenotype. We present a web-based tool (covid-omics.app) enabling interactive exploration of our compendium and illustrate its utility through a machine learning approach for prediction of COVID-19 severity.
https://www.sciencedirect.com/science/article/pii/S2405471220303719?via%3Dihub
https://covid-omics.app:8080/
key: 数据集:covid-19 阳性和阴性病人的128份血液样本进行RNA-seq and high-resolution mass spectrometry
跨组学 鉴定与疾病严重程度相关的分子特征 219 molecular features 表明在COVID-19下确实可以调节关键的生物学过程,包括补体系统激活,脂质转运,血管损伤,血小板激活和脱粒,凝血,和急性期反应 我们还提供了一个应用示例,该示例利用此资源基于所有组学数据开发疾病严重性预测模型


Abstract
Motivation
Gene network inference and master regulator analysis (MRA) have been widely adopted to define specific transcriptional perturbations from gene expression signatures. Several tools exist to perform such analyses but most require a computer cluster or large amounts of RAM to be executed.
Results
We developed corto, a fast and lightweight R package to infer gene networks and perform MRA from gene expression data, with optional corrections for copy-number variations and able to run on signatures generated from RNA-Seq or ATAC-Seq data. We extensively benchmarked it to infer context-specific gene networks in 39 human tumor and 27 normal tissue datasets.
key:
1.基因网络推断
2.copy number变异校正
3.mra
4.网络富集可视化

Abstract
An updated Lnc2Cancer 3.0 (http://www.bio-bigdata.net/lnc2cancer or http://bio-bigdata.hrbmu.edu.cn/lnc2cancer) database, which includes comprehensive data on experimentally supported long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs) associated with human cancers. In addition, web tools for analyzing lncRNA expression by high-throughput RNA sequencing (RNA-seq) and single-cell RNA-seq (scRNA-seq) are described. Lnc2Cancer 3.0 was updated with several new features, including (i) Increased cancer-associated lncRNA entries over the previous version. The current release includes 9254 lncRNA-cancer associations, with 2659 lncRNAs and 216 cancer subtypes. (ii) Newly adding 1049 experimentally supported circRNA-cancer associations, with 743 circRNAs and 70 cancer subtypes. (iii) Experimentally supported regulatory mechanisms of cancer-related lncRNAs and circRNAs, involving microRNAs, transcription factors (TF), genetic variants, methylation and enhancers were included. (iv) Appending experimentally supported biological functions of cancer-related lncRNAs and circRNAs including cell growth, apoptosis, autophagy, epithelial mesenchymal transformation (EMT), immunity and coding ability. (v) Experimentally supported clinical relevance of cancer-related lncRNAs and circRNAs in metastasis, recurrence, circulation, drug resistance, and prognosis was included. Additionally, two flexible online tools, including RNA-seq and scRNA-seq web tools, were developed to enable fast and customizable analysis and visualization of lncRNAs in cancers. Lnc2Cancer 3.0 is a valuable resource for elucidating the associations between lncRNA, circRNA and cancer.
key:
lncRNA和circRNA在癌症相关调控机制中的作用,包括增强子,遗传变异,microRNA(miRNA)相互作用,转录因子(TFs)和甲基化修饰
webtools: 通过RNA-seq 和scRNA分析lncRNA的表达量

Abstract
There is an urgent need to better understand the pathophysiology of Coronavirus disease 2019 (COVID-19), the global pandemic caused by SARS-CoV-2, which has infected more than three million people worldwide1. Approximately 20% of patients with COVID-19 develop severe disease and 5% of patients require intensive care2. Severe disease has been associated with changes in peripheral immune activity, including increased levels of pro-inflammatory cytokines3,4 that may be produced by a subset of inflammatory monocytes5,6, lymphopenia7,8 and T cell exhaustion9,10. To elucidate pathways in peripheral immune cells that might lead to immunopathology or protective immunity in severe COVID-19, we applied single-cell RNA sequencing (scRNA-seq) to profile peripheral blood mononuclear cells (PBMCs) from seven patients hospitalized for COVID-19, four of whom had acute respiratory distress syndrome, and six healthy controls. We identify reconfiguration of peripheral immune cell phenotype in COVID-19, including a heterogeneous interferon-stimulated gene signature, HLA class II downregulation and a developing neutrophil population that appears closely related to plasmablasts appearing in patients with acute respiratory failure requiring mechanical ventilation. Importantly, we found that peripheral monocytes and lymphocytes do not express substantial amounts of pro-inflammatory cytokines. Collectively, we provide a cell atlas of the peripheral immune response to severe COVID-19.
https://www.nature.com/articles/s41591-020-0944-y
key:
7个Covid-19 住院患者,其中4个患有急性呼吸窘迫综合症。6个健康对照的外周血单核细胞测序数据。
做了哪些分析?
1.重型COVID-19 的外周免疫细胞的单细胞转录图谱
主成分分析,细胞聚类,表型差异
2.量化COVID-19驱动的细胞类型比例变化以及全新中性粒细胞亚群的发现
3.对单核细胞进行进一步降维分析
4.确定COVID-19样本中引起免疫细胞表型变化的基因,HLA II下调, 干扰素刺激基因异质性
5.对COVID-19样本中T细胞及NK细胞的分析以及浆母细胞与中性粒细胞表型连续性的发现

Abstract
Viruses are a constant threat to global health as highlighted by the current COVID-19 pandemic. Currently, lack of data underlying how the human host interacts with viruses, including the SARS-CoV-2 virus, limits effective therapeutic intervention. We introduce Viral-Track, a computational method that globally scans unmapped single-cell RNA sequencing (scRNA-seq) data for the presence of viral RNA, enabling transcriptional cell sorting of infected versus bystander cells. We demonstrate the sensitivity and specificity of Viral-Track to systematically detect viruses from multiple models of infection, including hepatitis B virus, in an unsupervised manner. Applying Viral-Track to bronchoalveloar-lavage samples from severe and mild COVID-19 patients reveals a dramatic impact of the virus on the immune system of severe patients compared to mild cases. Viral-Track detects an unexpected co-infection of the human metapneumovirus, present mainly in monocytes perturbed in type-I interferon (IFN)-signaling. Viral-Track provides a robust technology for dissecting the mechanisms of viral-infection and pathology.
key:
病毒追踪,扫描scRNA-seq数据是否存在病毒RNA,从而对感染细胞和未感染细胞进行分类。
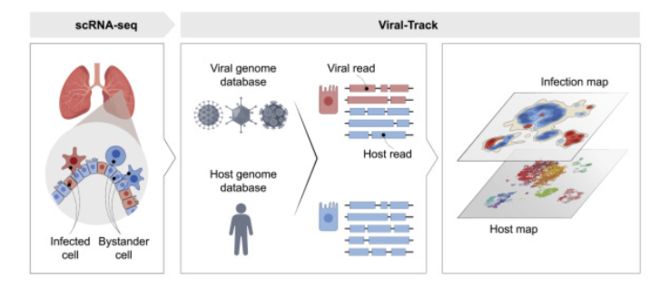

Summary
Diabetes is associated with increased mortality from severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). Given literature suggesting a potential association between SARS-CoV-2 infection and diabetes induction, we examined pancreatic expression of angiotensin-converting enzyme 2 (ACE2), the key entry factor for SARS-CoV-2 infection. Specifically, we analyzed five public scRNA-seq pancreas datasets and performed fluorescence in situ hybridization, western blotting, and immunolocalization for ACE2 with extensive reagent validation on normal human pancreatic tissues across the lifespan, as well as those from coronavirus disease 2019 (COVID-19) cases. These in silico and ex vivo analyses demonstrated prominent expression of ACE2 in pancreatic ductal epithelium and microvasculature, but we found rare endocrine cell expression at the mRNA level. Pancreata from individuals with COVID-19 demonstrated multiple thrombotic lesions with SARS-CoV-2 nucleocapsid protein expression that was primarily limited to ducts. These results suggest SARS-CoV-2 infection of pancreatic endocrine cells, via ACE2, is an unlikely central pathogenic feature of COVID-19-related diabetes.

key: 糖尿病与COVID-19高死亡率相关,探索这种关联。
ACE2是SARS-COV-2感染的关键进入因素。
数据:正常胰腺组织和感染COVID-19的胰腺组织。ACE2在胰腺导管上皮和微脉管系统中有突出表达,但我们发现在mRNA水平上罕见的内分泌细胞表达。来自患有COVID-19的个体的胰腺表现出多处血栓性病变,其SARS-CoV-2核衣壳蛋白表达主要限于导管。这些结果表明,通过ACE2感染胰腺内分泌细胞的SARS-CoV-2是COVID-19相关糖尿病的不太可能的中央致病特征。

Abstract
Single-cell RNA sequencing (scRNA-seq) technologies allow researchers to uncover the biological states of a single cell at high resolution. For computational efficiency and easy visualization, dimensionality reduction is necessary to capture gene expression patterns in low-dimensional space. Here we propose an ensemble method for simultaneous dimensionality reduction and feature gene extraction (EDGE) of scRNA-seq data. Different from existing dimensionality reduction techniques, the proposed method implements an ensemble learning scheme that utilizes massive weak learners for an accurate similarity search. Based on the similarity matrix constructed by those weak learners, the low-dimensional embedding of the data is estimated and optimized through spectral embedding and stochastic gradient descent. Comprehensive simulation and empirical studies show that EDGE is well suited for searching for meaningful organization of cells, detecting rare cell types, and identifying essential feature genes associated with certain cell types.
key:
单细胞数据的降维和特征提取
学习细胞之间的相似性