前言
在利用cellranger比对单细胞reads时,可以发现有STAR的进程夹杂在里面,那么STAR可以用来比对单细胞数据吗?在STAR的2.7版本中(2.7.6a)出现了STARsolo,可以进行单细胞数据的比对,由此可见STAR的强大
Cellranger输出结果
在使用STAR之前,先看一下cellranger的输出结果
.
├── analysis
│ ├── clustering
│ ├── diffexp
│ ├── pca
│ ├── tsne
│ └── umap
├── cloupe.cloupe
├── filtered_feature_bc_matrix
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── filtered_feature_bc_matrix.h5
├── metrics_summary.csv
├── molecule_info.h5
├── possorted_genome_bam.bam
├── possorted_genome_bam.bam.bai
├── raw_feature_bc_matrix
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── raw_feature_bc_matrix.h5
└── web_summary.html
为方便查看,cellranger提供了一个网页端的结果,我们主要观察细胞和基因数目的评估即可,后续的聚类工作由seurat完成
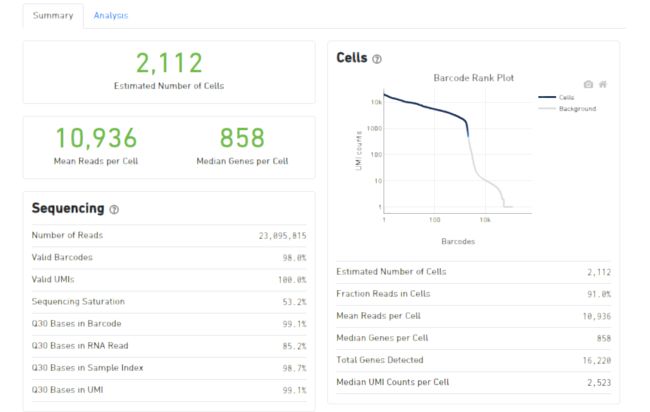
在结果目录,可以看到如下两个目录
raw_feature_bc_matrix
filtered_gene_bc_matrices
raw目录下是所有的barcode信息,包含了细胞相关的barcoed和背景barcode,而filter目录下只包含细胞相关的barcode信息,内容如下
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
后缀为mtx的文件记录的就是基因的表达量信息,可以导入R或者python中查看,barcodes相当于一个细胞,features代表不同的基因,barcodes文件在STARsolo中会用到,这就是为什么我要先说明一下cellranger的输出结果
利用STAR分析10X数据
STARsolo被设计为替代10X CellRanger基因定量比对软件。而且STARsolo的速度是cellranger的十倍(具体怎么样我也不知道)
建立索引:
STAR --runMode genomeGenerate --genomeDir ghg38/ --genomeFastaFiles Homo_sapiens.GRCh38.dna.primary_assembly.fa --sjdbGTFfile Homo_sapiens.GRCh38.93.filtered.gtf
其实利用cellranger构建的索引原则上也能用,在GRCh38/star/下,但是由于STAR版本问题可能会导致无法识别,因为cellranger用的STAR构建的索引和我们自己用的STAR版本是不一致的。
STARsolo与普通的转录组比对区别在于你需要在比对时加上whitelist,whitelist文件格式在10X官网有写,我们可以利用cellranger的barcodes.tsv.gz文件获得
zcat barcodes.tsv.gz>whitelist
sed -i "s\-1\\g" whitelist
需要注意ReadFilesIn先读入测序数据,再读入barcode+UMI文件,即先读入R2再读入R1
STAR --genomeDir ghg38/ --readFilesCommand zcat --readFilesIn SRR7722939/SRR7722939_S1_L001_R2_001.fastq.gz SRR7722939/SRR7722939_S1_L001_R1_001.fastq.gz --soloType CB_UMI_Simple --soloCBwhitelist whitelist --runThreadN 8
结果默认保存在Solo.out文件中,8线程只用了10min左右,确实要快一点
├── Barcodes.stats
└── Gene
├── Features.stats
├── filtered
│ ├── barcodes.tsv
│ ├── features.tsv
│ └── matrix.mtx
├── raw
│ ├── barcodes.tsv
│ ├── features.tsv
│ └── matrix.mtx
├── Summary.csv
└── UMIperCellSorted.txt
看一下Summary里是啥
Number of Reads,23095815
Reads With Valid Barcodes,0.979732
Sequencing Saturation,0.529367
Q30 Bases in CB+UMI,0.991351
Q30 Bases in RNA read,0.842989
Reads Mapped to Genome: Unique+Multiple,0.959769
Reads Mapped to Genome: Unique,0.874368
Reads Mapped to Transcriptome: Unique+Multipe Genes,0.63797
Reads Mapped to Transcriptome: Unique Genes,0.613655
Estimated Number of Cells,2048
Reads in Cells Mapped to Unique Genes,12815372
Fraction of Reads in Cells,0.904219
Mean Reads per Cell,6257
Median Reads per Cell,5544
UMIs in Cells,5978718
Mean UMI per Cell,2919
Median UMI per Cell,2577
Mean Genes per Cell,915
Median Genes per Cell,873
Total Genes Detected,16265
可以看到比起cellranger,STAR捕获到的细胞数少一点,而且每个细胞的reads要低一点,其他差不多,后续将使用Seurat包对两组数据进行比较
STARsolo其他命令
--soloType
default: None
string(s): type of single-cell RNA-seq
CB_UMI_Simple
(a.k.a. Droplet) one UMI and one Cell Barcode of xed length in
read2, e.g. Drop-seq and 10X Chromium.
CB_UMI_Complex
one UMI of xed length, but multiple Cell Barcodes of varying length,
as well as adapters sequences are allowed in read2 only, e.g. inDrop.
CB_samTagOut
output Cell Barcode as CR and/or CB SAm tag. No UMI counting.
{readFilesIn cDNA read1 [cDNA read2 if paired-end]
CellBarcode read . Requires {outSAMtype BAM Unsorted [and/or
SortedByCoordinate]
SmartSeq
Smart-seq: each cell in a separate FASTQ (paired- or single-end),
barcodes are corresponding read-groups, no UMI sequences,
alignments deduplicated according to alignment start and end (after
extending soft-clipped bases)
--soloCBwhitelist
default: -
string(s): le(s) with whitelist(s) of cell barcodes. Only {soloType
CB UMI Complex allows more than one whitelist le.
None
no whitelist: all cell barcodes are allowed
--soloCBstart
default: 1
int>0: cell barcode start base
--soloCBlen
default: 16
int>0: cell barcode length
--soloUMIstart
default: 17
int>0: UMI start base
--soloUMIlen
default: 10
int>0: UMI length
--soloBarcodeReadLength
default: 1
int: length of the barcode read
1
equal to sum of soloCBlen+soloUMIlen
0
not dened, do not check
--soloCBposition
default: -
strings(s) position of Cell Barcode(s) on the barcode read.
Presently only works with {soloType CB UMI Complex, and barcodes are
assumed to be on Read2.
Format for each barcode: startAnchor startPosition endAnchor endPosition
start(end)Anchor denes the Anchor Base for the CB: 0: read start; 1: read
end; 2: adapter start; 3: adapter end
start(end)Position is the 0-based position with of the CB start(end) with
respect to the Anchor Base
String for di�erent barcodes are separated by space.
Example: inDrop (Zilionis et al, Nat. Protocols, 2017):
{soloCBposition 0 0 2 -1 3 1 3 8
--soloUMIposition
default: -
string position of the UMI on the barcode read, same as soloCBposition
--soloAdapterSequence
default: -
string: adapter sequence to anchor barcodes.
--soloAdapterMismatchesNmax
default: 1
int>0: maximum number of mismatches allowed in adapter sequence.
--soloCBmatchWLtype
default: 1MM multi
string: matching the Cell Barcodes to the WhiteList
Exact
only exact matches allowed
1MM
only one match in whitelist with 1 mismatched base allowed. Allowed
CBs have to have at least one read with exact match.
1MM_multi
multiple matches in whitelist with 1 mismatched base allowed,
posterior probability calculation is used choose one of the matches.
Allowed CBs have to have at least one read with exact match. Similar to
CellRanger 2.2.0
1MM_multi_pseudocounts
same as 1MM Multi, but pseudocounts of 1 are added to all whitelist
barcodes.
Similar to CellRanger 3.x.x
--soloStrand
default: Forward
string: strandedness of the solo libraries:
Unstranded
no strand information
Forward
read strand same as the original RNA molecule
Reverse
read strand opposite to the original RNA molecule
--soloFeatures
default: Gene
string(s): genomic features for which the UMI counts per Cell Barcode are
collected
Gene
genes: reads match the gene transcript
SJ
splice junctions: reported in SJ.out.tab
GeneFull
full genes: count all reads overlapping genes' exons and introns
--soloUMIdedup
default: 1MM_All
string(s): type of UMI deduplication (collapsing) algorithm
1MM_All
all UMIs with 1 mismatch distance to each other are collapsed (i.e.
counted once)
1MM_Directional
follows the "directional" method from the UMI-tools by Smith, Heger
and Sudbery (Genome Research 2017).
Exact
only exactly matching UMIs are collapsed
NoDedup
no deduplication of UMIs, count all reads. Allowed for --soloType
SmartSeq
--soloUMIfiltering
default: -
string(s) type of UMI ltering
-
basic ltering: remove UMIs with N and homopolymers (similar to
CellRanger 2.2.0)
MultiGeneUMI
remove lower-count UMIs that map to more than one gene
(introduced in CellRanger 3.x.x)
--soloOutFileNames
default: Solo.out/ features.tsv barcodes.tsv matrix.mtx
string(s) le names for STARsolo output:
le name prex gene names barcode sequences cell feature count matrix
--soloCellFilter
default: CellRanger2.2 3000 0.99 10
string(s): cell ltering type and parameters
CellRanger2.2
simple ltering of CellRanger 2.2, followed by three numbers: number
of expected cells, robust maximum percentile for UMI count,
maximum to minimum ratio for UMI count
TopCells
only report top cells by UMI count, followed by the exact number of
cells
None
do not output ltered cells
--soloOutFormatFeaturesGeneField3
default: "Gene Expression"
string(s): eld 3 in the Gene features.tsv le. If "-", then no 3rd eld is output.
转载请注明:周小钊的博客>>>单细胞实战(3):STAR分析单细胞数据