跟着 Nat Med. 学作图 | GSVA+limma差异通路分析+发散条形图
跟着 Nat Med. 学作图 | GSVA+limma差异通路分析+发散条形图

Lambrechts D, Wauters E, Boeckx B, et al. Phenotype molding of stromal cells in the lung tumor microenvironment[J]. Nat Med, 2018, 24(8): p. 1277-1289.
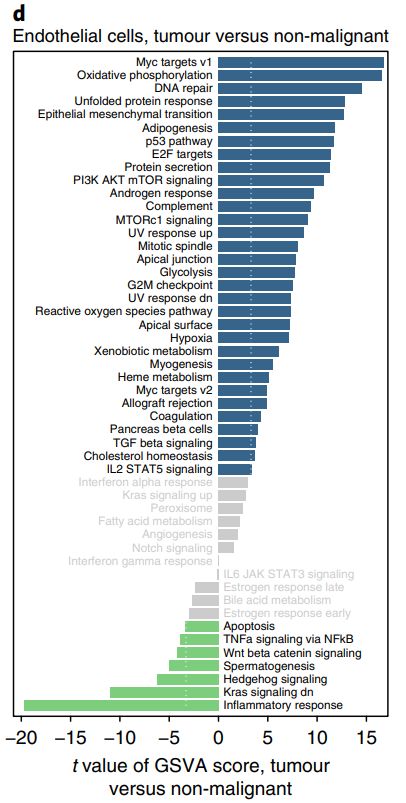
最近很多同学在后台说要讲一下这个图,今天来简单写一下。
Gene Set Variation Analysis(GSVA)被称为基因集变异分析,是一种非参数的无监督分析方法,主要用来评估芯片和转录组的基因集富集结果。主要是通过将基因在不同样品间的表达量矩阵转化成基因集在样品间的表达量矩阵,从而来评估不同的代谢通路在不同样品间是否富集。通常用于单细胞转录组中,不同细胞类型的差异基因集的比较。
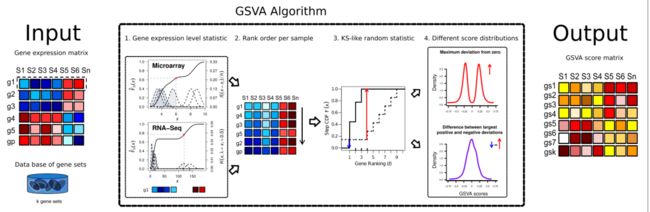 GSVA流程示意图。doi:10.1186/1471-2105-14-7
GSVA流程示意图。doi:10.1186/1471-2105-14-7
示例数据及代码领取
赞赏
10¥或点赞在看本推文并转发至朋友圈集赞5个。木舟笔记
2022年VIP会员可以免费领取。关于木舟笔记2022年度VIP会员企划
权益:
1. 2022年度木舟笔记所有推文示例数据及代码(含21年,除Cell合集外资源。 2. 木舟笔记科研交流群。 3. 半价购买`跟着Cell学作图系列合集`[(免费教程+代码领取)|跟着Cell学作图系列合集](http://mp.weixin.qq.com/s?__biz=MzIxMDExNDE0OQ==&mid=2247486072&idx=1&sn=d579a62420f722285eac318c4114d199&chksm=9768c972a01f4064a84f30fd48b8c2107af543c31b6adb5f36234b1377ac42c8af54f45efda9#rd)。收费:
99¥/人。可添加微信:
mzbj0002转账,或直接在文末打赏。
GSVA
导入示例数据
## 为正常和肿瘤的内皮细胞的基因表达矩阵
library(readxl)
library(dplyr)
dat <- read_excel("data_test.xlsx")
dat <- dat %>% data.frame()
row.names(dat) <- dat$gene
dat <- dat[,-1]
head(dat)> head(dat)
EC1_T EC2_T EC3_T EC0_N EC1_N EC3_N EC4_N
RP11-34P13.3 0 0 0 0 0 0 0
FAM138A 0 0 0 0 0 0 0
OR4F5 0 0 0 0 0 0 0
RP11-34P13.7 0 0 0 0 0 0 0
RP11-34P13.8 0 0 0 0 0 0 0
RP11-34P13.14 0 0 0 0 0 0 0参考基因集数据库
MSigDB数据库具体介绍详见:Q&A | 如何使用clusterProfiler对MSigDB数据库进行富集分析
这里我们使用:hallmark gene sets
开始分析
#BiocManager::install("GSEABase")
BiocManager::install("GSVA", version = "3.14") # R 4.1.2 注意版本w
library('GSEABase')
library(GSVA)
geneSets <- getGmt('h.all.v7.5.1.symbols.gmt') ###下载的基因集
GSVA_hall <- gsva(expr=as.matrix(dat),
gset.idx.list=geneSets,
mx.diff=T, # 数据为正态分布则T,双峰则F
kcdf="Gaussian", #CPM, RPKM, TPM数据就用默认值"Gaussian", read count数据则为"Poisson",
parallel.sz=4) # 并行线程数目
head(GSVA_hall)> head(GSVA_hall)
EC1_T EC2_T EC3_T EC0_N
HALLMARK_TNFA_SIGNALING_VIA_NFKB -0.43490528 -0.36929145 -0.4694267 0.57790329
HALLMARK_HYPOXIA -0.19830871 -0.15040810 -0.1237437 0.31595670
HALLMARK_CHOLESTEROL_HOMEOSTASIS -0.15223695 -0.15160770 -0.0505465 0.24273366
HALLMARK_MITOTIC_SPINDLE -0.29757233 0.20140000 0.3185932 -0.09284047
HALLMARK_WNT_BETA_CATENIN_SIGNALING -0.08827878 0.03086390 0.2383694 0.24045204
HALLMARK_TGF_BETA_SIGNALING -0.26357054 -0.01068719 0.3041132 0.28761931
EC1_N EC3_N EC4_N
HALLMARK_TNFA_SIGNALING_VIA_NFKB -0.1673606 0.2198982 0.33940521
HALLMARK_HYPOXIA -0.3292568 0.0906295 0.01407149
HALLMARK_CHOLESTEROL_HOMEOSTASIS -0.3566404 0.1532081 0.05151732
HALLMARK_MITOTIC_SPINDLE -0.3673806 0.1091566 -0.15295077
HALLMARK_WNT_BETA_CATENIN_SIGNALING -0.2352282 -0.1027337 -0.15598311
HALLMARK_TGF_BETA_SIGNALING -0.4387025 0.2336080 -0.14090342limma差异通路分析
## limma
#BiocManager::install('limma')
library(limma)
# 设置或导入分组
group <- factor(c(rep("Tumor", 3), rep("Normal", 4)), levels = c('Tumor', 'Normal'))
design <- model.matrix(~0+group)
colnames(design) = levels(factor(group))
rownames(design) = colnames(GSVA_hall)
design
# Tunor VS Normal
compare <- makeContrasts(Tumor - Normal, levels=design)
fit <- lmFit(GSVA_hall, design)
fit2 <- contrasts.fit(fit, compare)
fit3 <- eBayes(fit2)
Diff <- topTable(fit3, coef=1, number=200)
head(Diff)> head(Diff)
logFC AveExpr t P.Value
HALLMARK_INTERFERON_GAMMA_RESPONSE -0.7080628 -0.021269395 -4.393867 0.0002713325
HALLMARK_INTERFERON_ALPHA_RESPONSE -0.7419587 -0.044864170 -4.299840 0.0003385128
HALLMARK_INFLAMMATORY_RESPONSE -0.5940473 -0.003525139 -4.193096 0.0004352397
HALLMARK_IL6_JAK_STAT3_SIGNALING -0.6117943 -0.038379008 -4.157977 0.0004727716
HALLMARK_TNFA_SIGNALING_VIA_NFKB -0.6670027 -0.043396767 -4.149307 0.0004825257
HALLMARK_ALLOGRAFT_REJECTION -0.4645747 0.015248663 -3.278981 0.0036971108
adj.P.Val B
HALLMARK_INTERFERON_GAMMA_RESPONSE 0.004825257 0.43891329
HALLMARK_INTERFERON_ALPHA_RESPONSE 0.004825257 0.22763343
HALLMARK_INFLAMMATORY_RESPONSE 0.004825257 -0.01221232
HALLMARK_IL6_JAK_STAT3_SIGNALING 0.004825257 -0.09109393
HALLMARK_TNFA_SIGNALING_VIA_NFKB 0.004825257 -0.11056468
HALLMARK_ALLOGRAFT_REJECTION 0.030809257 -2.03863951发散条形图绘制
## barplot
dat_plot <- data.frame(id = row.names(Diff),
t = Diff$t)
# 去掉"HALLMARK_"
library(stringr)
dat_plot$id <- str_replace(dat_plot$id , "HALLMARK_","")
# 新增一列 根据t阈值分类
dat_plot$threshold = factor(ifelse(dat_plot$t >-2, ifelse(dat_plot$t >= 2 ,'Up','NoSignifi'),'Down'),levels=c('Up','Down','NoSignifi'))
# 排序
dat_plot <- dat_plot %>% arrange(t)
# 变成因子类型
dat_plot$id <- factor(dat_plot$id,levels = dat_plot$id)
# 绘制
library(ggplot2)
library(ggtheme)
# install.packages("ggprism")
library(ggprism)
p <- ggplot(data = dat_plot,aes(x = id,y = t,fill = threshold)) +
geom_col()+
coord_flip() +
scale_fill_manual(values = c('Up'= '#36638a','NoSignifi'='#cccccc','Down'='#7bcd7b')) +
geom_hline(yintercept = c(-2,2),color = 'white',size = 0.5,lty='dashed') +
xlab('') +
ylab('t value of GSVA score, tumour versus non-malignant') + #注意坐标轴旋转了
guides(fill=F)+ # 不显示图例
theme_prism(border = T) +
theme(
axis.text.y = element_blank(),
axis.ticks.y = element_blank()
)
p
# 添加标签
# 此处参考了:https://mp.weixin.qq.com/s/eCMwWCnjTyQvNX2wNaDYXg
# 小于-2的数量
low1 <- dat_plot %>% filter(t < -2) %>% nrow()
# 小于0总数量
low0 <- dat_plot %>% filter( t < 0) %>% nrow()
# 小于2总数量
high0 <- dat_plot %>% filter(t < 2) %>% nrow()
# 总的柱子数量
high1 <- nrow(dat_plot)
# 依次从下到上添加标签
p <- p + geom_text(data = dat_plot[1:low1,],aes(x = id,y = 0.1,label = id),
hjust = 0,color = 'black') + # 小于-1的为黑色标签
geom_text(data = dat_plot[(low1 +1):low0,],aes(x = id,y = 0.1,label = id),
hjust = 0,color = 'grey') + # 灰色标签
geom_text(data = dat_plot[(low0 + 1):high0,],aes(x = id,y = -0.1,label = id),
hjust = 1,color = 'grey') + # 灰色标签
geom_text(data = dat_plot[(high0 +1):high1,],aes(x = id,y = -0.1,label = id),
hjust = 1,color = 'black') # 大于1的为黑色标签
ggsave("gsva_bar.pdf",p,width = 8,height = 8)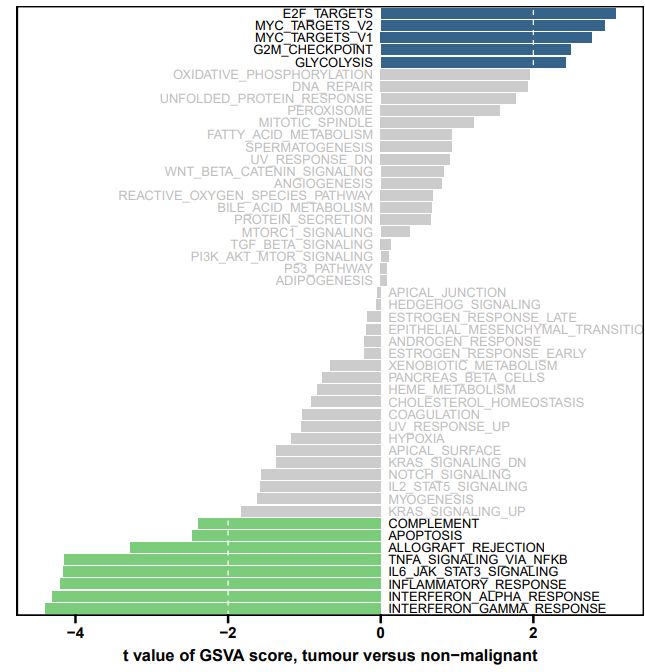 结果展示
结果展示
往期
跟着Cell学作图 | Proteomaps图
- END -
