SCS【4】单细胞转录组数据可视化分析 (Seurat 4.0)

点击关注,桓峰基因
桓峰基因的教程不但教您怎么使用,还会定期分析一些相关的文章,学会教程只是基础,但是如果把分析结果整合到文章里面才是目的,觉得我们这些教程还不错,并且您按照我们的教程分析出来不错的结果发了文章记得告知我们,并在文章中感谢一下我们哦!
公司英文名称:Kyoho Gene Technology (Beijing) Co.,Ltd.
桓峰基因公众号推出单细胞系列教程,有需要生信的老师可以联系我们!首选看下转录分析教程整理如下:
Topic 6. 克隆进化之 Canopy
Topic 7. 克隆进化之 Cardelino
Topic 8. 克隆进化之 RobustClone
SCS【1】今天开启单细胞之旅,述说单细胞测序的前世今生
SCS【2】单细胞转录组 之 cellranger
SCS【3】单细胞转录组数据 GEO 下载及读取
SCS【4】单细胞转录组数据可视化分析 (Seurat 4.0)
今天来说说单细胞转录组数据的分析细节,学会这些分析结果,距离发文章就只差样本的选择了,有创新性的样本将成为文章的亮点,并不是分析内容了!
前 言
在本教程中,我们将分析免费从10X基因组公司获得的外周血单个核细胞(PBMC)数据集。Illumina NextSeq 500对2700个单细胞进行了测序的原始数据。
我们从读入数据开始。Read10X()函数从10X读取cellranger管道的输出,返回一个唯一的分子标识(UMI)计数矩阵。这个矩阵中的值表示每个特征(即基因;行),在每个单元格(列)中检测到。
接下来我们使用计数矩阵来创建一个Seurat对象。该对象作为一个容器,包含单格数据集的数据(如计数矩阵)和分析(如PCA或聚类结果)。
官网教程如下:
https://satijalab.org/seurat/articles/pbmc3k_tutorial.html
软件安装
整个过程需要使用的软件包 Seurat ,其他一些辅助性数据出来的软件包:dplyr,patchwork等。
if(!require(Seurat))
BiocManager::install("Seurat")
数据读取
下载数据后利用Read10X对数据进行读取即可。数据下载地址:
https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
下载之后我们就可以看到文件夹里面只有三个文件,跟我们之前介绍GEO下载的数据是一致的:
-
barcodes.tsv
-
genes.tsv
-
matrix.mtx
library(Seurat)
### Setup the Seurat Object Load the PBMC dataset
pbmc.data <- Read10X(data.dir = "./filtered_gene_bc_matrices/hg19")
# Initialize the Seurat object with the raw (non-normalized data).
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3,
min.features = 200)
pbmc
## An object of class Seurat
## 13714 features across 2700 samples within 1 assay
## Active assay: RNA (13714 features, 0 variable features)
#### QC and selecting cells for further analysis The [[ operator can add
#### columns to object metadata. This is a great place to stash QC stats
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
# Visualize QC metrics as a violin plot
我们可以看一下数据的结构以及大小等信息
# Lets examine a few genes in the first thirty cells
pbmc.data[c("CD3D", "TCL1A", "MS4A1"), 1:30]
## 3 x 30 sparse Matrix of class "dgCMatrix"
##
## CD3D 4 . 10 . . 1 2 3 1 . . 2 7 1 . . 1 3 . 2 3 . . . . . 3 4 1 5
## TCL1A . . . . . . . . 1 . . . . . . . . . . . . 1 . . . . . . . .
## MS4A1 . 6 . . . . . . 1 1 1 . . . . . . . . . 36 1 2 . . 2 . . . .
dense.size <- object.size(as.matrix(pbmc.data))
dense.size
## 709591472 bytes
sparse.size <- object.size(pbmc.data)
sparse.size
## 29905192 bytes
dense.size/sparse.size
## 23.7 bytes
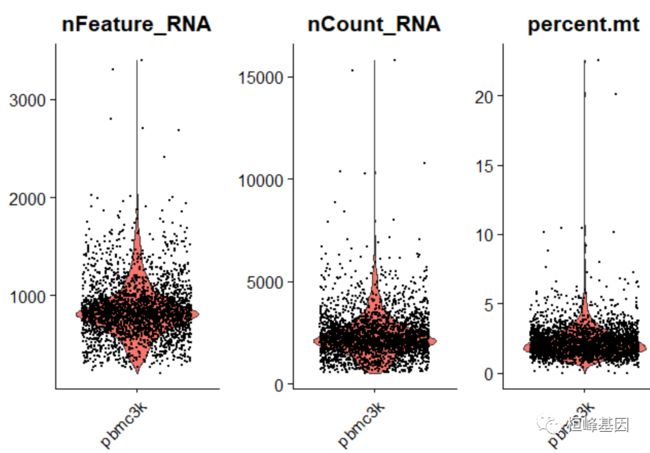
在下面的示例中,我们可视化QC指标,并使用这些指标过滤单元格。
-
过滤具有独特特性的单元格,其计数超过2500或小于200
-
过滤那些线粒体数量为5%的细胞
将QC指标可视化绘制小提琴图
# Visualize QC metrics as a violin plot
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
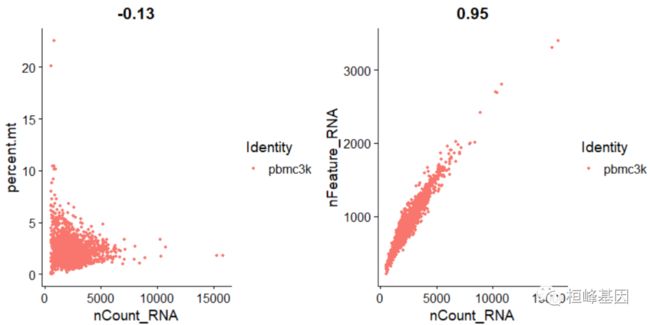
FeatureScatter通常用于可视化特征-特征关系,但也可以用于对象计算的任何东西,例如对象元数据中的列,PC分数等。
# FeatureScatter is typically used to visualize feature-feature relationships,
# but can be used for anything calculated by the object, i.e. columns in object
# metadata, PC scores etc.
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt <
5)
实例操作
1. 标准化分析
在从数据集中删除不需要的单元格之后,下一步是规范化数据。默认情况下,我们使用全局缩放规范化方法“LogNormalize”,该方法将每个单元格的特征表达式度量按总表达式进行规范化,并将其乘以一个缩放因子(默认为10,000),然后对结果进行日志转换。归一化的值存储在pbmc[[“RNA”]]@data中。
################ Normalizing the data
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
为了清晰起见,在前面这行代码(以及以后的命令)中,我们为函数调用中的某些参数提供了默认值。然而,这不是必需的,相同的行为可以通过以下方式实现:
pbmc <- NormalizeData(pbmc)
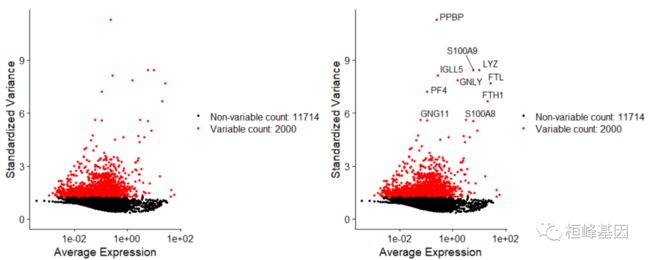
2. 高频变异基因识别(基因选择)
我们计算数据集中表现出高细胞间变异的特征子集(即,它们在一些细胞中高表达,在另一些细胞中低表达)。我们和其他人已经发现,在下游分析中关注这些基因有助于突出单细胞数据集中的生物信号。
我们在Seurat中的过程在这里进行了详细描述,并通过直接建模单格数据中固有的均值-方差关系对以前的版本进行了改进,并在FindVariableFeatures()函数中实现。默认情况下,我们为每个数据集返回2000个特性。这些将用于下游分析,如PCA。
####Identification of highly variable features (feature selection)
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
# Identify the 10 most highly variable genes
top10 <- head(VariableFeatures(pbmc), 10)
# plot variable features with and without labels
plot1 <- VariableFeaturePlot(pbmc)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
plot1 + plot2
接下来,我们应用线性变换(“缩放”),这是在PCA等降维技术之前的一个标准预处理步骤。ScaleData()函数:
-
改变每个基因的表达,使细胞间的平均表达为0
-
衡量每个基因的表达,使细胞间的方差为1
-
这一步在下游分析中给予同等的权重,因此高表达基因不会占主导地位
结果存储在pbmc[[“RNA”]]@scale.data中
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, features = all.genes)
缩放是Seurat工作流程中的一个重要步骤,但仅适用于将被用作PCA输入的基因。因此,ScaleData()中的默认情况是只对前面确定的变量特性执行伸缩(默认为2000)。为此,请忽略前面函数调用中的features参数。
pbmc <- ScaleData(pbmc)
PCA和聚类结果将不受影响。然而,Seurat热图(使用DoHeatmap()生成,如下图所示)需要对热图中的基因进行缩放,以确保高表达基因不会主导热图。为了确保我们不会在之后的热图中遗漏任何基因,我们将在本教程中缩放所有基因。
线性降维
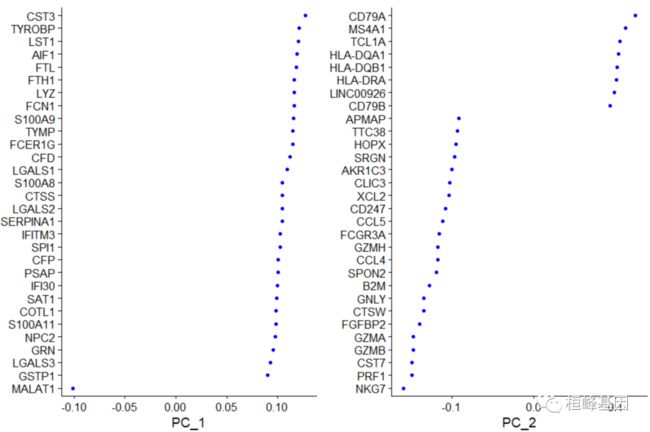
接下来,我们对缩放后的数据进行主成分分析。默认情况下,只有之前确定的变量特性被用作输入,但是如果您希望选择一个不同的子集,可以使用features参数来定义。Seurat提供了几种有用的方法来可视化单元格和定义PCA的特性,包括vizdimreduce (), DimPlot()和DimHeatmap()
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))
# Examine and visualize PCA results a few different ways
print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
## PC_ 1
## Positive: CST3, TYROBP, LST1, AIF1, FTL
## Negative: MALAT1, LTB, IL32, IL7R, CD2
## PC_ 2
## Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1
## Negative: NKG7, PRF1, CST7, GZMB, GZMA
## PC_ 3
## Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPB1
## Negative: PPBP, PF4, SDPR, SPARC, GNG11
## PC_ 4
## Positive: HLA-DQA1, CD79B, CD79A, MS4A1, HLA-DQB1
## Negative: VIM, IL7R, S100A6, IL32, S100A8
## PC_ 5
## Positive: GZMB, NKG7, S100A8, FGFBP2, GNLY
## Negative: LTB, IL7R, CKB, VIM, MS4A7
VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")
维度聚类图
DimPlot(pbmc, reduction = "pca")
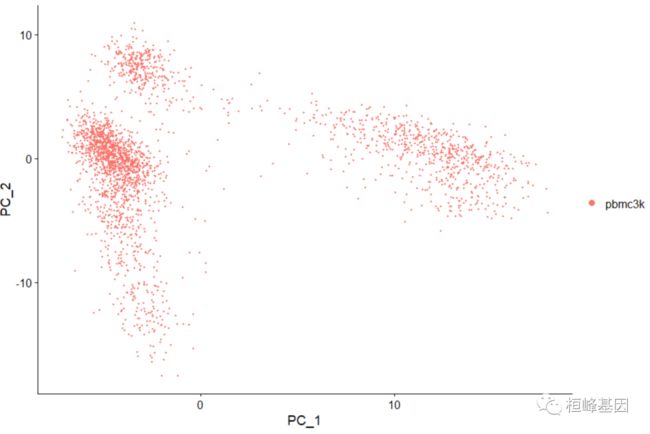
特别是,DimHeatmap()允许轻松地探索数据集中异构性的主要来源,并且在试图决定包含哪些pc进行进一步的下游分析时非常有用。根据PCA评分对细胞和特征进行排序。将单元格设置为一个数字,可以绘制光谱两端的“极端”单元格,这大大加快了绘制大型数据集的速度。虽然这是一个明显的监督分析,但我们发现这是一个有价值的工具,以探索相关的特征集。
DimHeatmap(pbmc, dims = 1, cells = 500, balanced = TRUE)
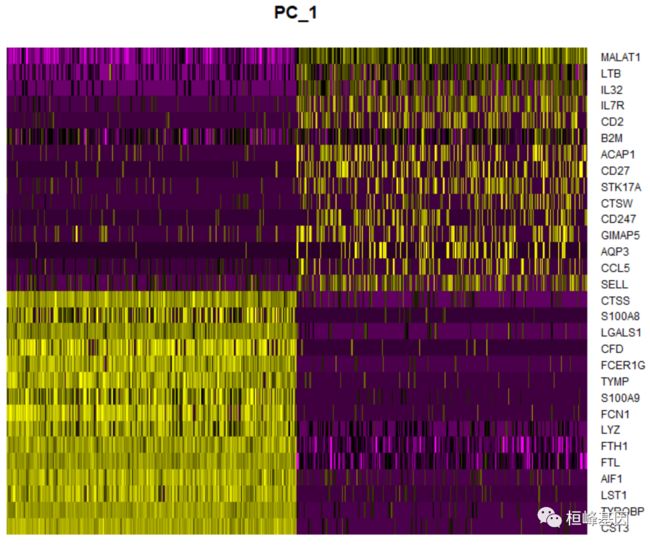
绘制多个热图
DimHeatmap(pbmc, dims = 1:15, cells = 500, balanced = TRUE)

确定数据集的“维度”
为了克服scRNA-seq数据中任何单一特征的大量技术噪声,Seurat基于它们的PCA分数聚类细胞,每个PC本质上代表一个“元特征”,该元特征结合了跨相关特征集的信息。因此,顶级主成分代表了数据集的鲁棒压缩。但是,我们应该选择包含多少个组件?10 ?20吗?100年?
在Macosko等人,我们实现了一个受JackStraw程序启发的重采样测试。我们随机排列数据的一个子集(默认为1%),并重新运行PCA,构建一个特征分数的“空分布”,并重复这个过程。我们认为“重要的”电脑是指那些具有较强的低p值特征的电脑。
# NOTE: This process can take a long time for big datasets, comment out for
# expediency. More approximate techniques such as those implemented in
# ElbowPlot() can be used to reduce computation time
pbmc <- JackStraw(pbmc, num.replicate = 100)
pbmc <- ScoreJackStraw(pbmc, dims = 1:20)
JackStrawPlot()函数提供了一个可视化工具,以比较每个PC的p值分布与均匀分布(虚线)。“重要的”pc会显示出较低的p值(虚线上方的实曲线)。在这种情况下,似乎有一个急剧下降的重要性。
JackStrawPlot(pbmc, dims = 1:15)
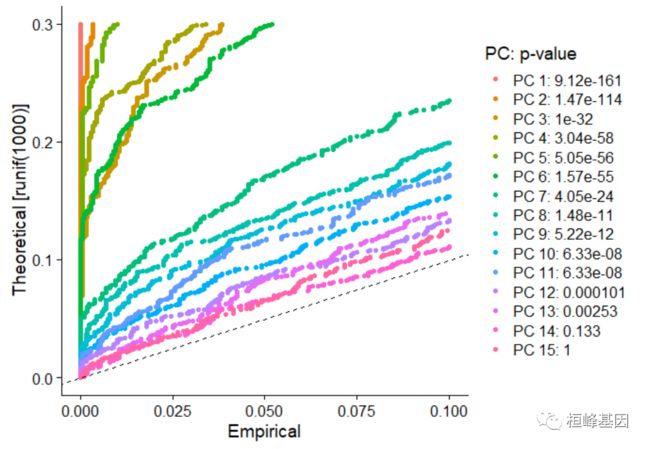
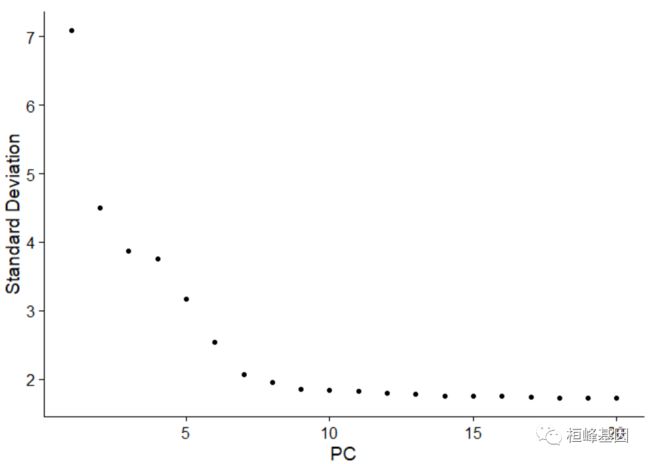
另一种启发式方法生成“肘部图”:基于每个元素解释的方差百分比对主要成分进行排序(肘部图()函数)。在这个例子中,我们可以观察到PC9-10周围有一个“弯头”,这表明大多数真实的信号被前10个pc捕获。
ElbowPlot(pbmc)
识别数据集的真实维度——对用户来说可能具有挑战性/不确定性。因此,我们建议考虑这三种方法。第一种是更有监督的,探索pc来确定异构性的相关来源,并可以与GS
EA结合使用。第二种方法基于一个随机的空模型实现一个统计检验,但是对于大型数据集来说是非常耗时的,并且可能不会返回一个明确的PC截断值。第三种是常用的启发式方法,可以立即计算出来。在这个例子中,所有三种方法都产生了类似的结果,但是我们可能会在PC 7-12之间选择任何一个作为截止点。
我们在这里选择了10个,但鼓励用户考虑以下几点:
树突状细胞和NK爱好者可能认识到,与pc12和pc13密切相关的基因定义了罕见的免疫亚群(即MZB1是浆细胞样dc的标记物)。然而,这些组非常罕见,在没有先验知识的情况下,它们很难从背景噪声中区分这种规模的数据集。我们鼓励用户使用不同数量的pc(10,15,甚至50!)重复下游分析。正如您将观察到的,结果通常不会有显著差异。我们建议用户在选择此参数时出错。例如,仅使用5个pc进行下游分析,会对结果产生显著的负面影响。
细胞聚类
Seurat v3应用了一种基于图的集群方法,建立在(Macosko等人)的初始策略之上。重要的是,驱动聚类分析的距离度量(基于先前确定的pc)保持不变。然而,我们将细胞距离矩阵划分为集群的方法有了显著的改进。我们的方法很大程度上受到了近期手稿的启发,这些手稿将基于图的聚类方法应用于scRNA-seq数据[SNN-Cliq, Xu和Su,生物信息学,2015]和CyTOF数据[表型,Levine等人,细胞,2015]。简单地说,这些方法将细胞嵌入到一个图结构中——例如一个k近邻(KNN)图,在具有相似特征表达模式的细胞之间绘制边缘,然后尝试将这个图划分为高度互联的“准小团体”或“社区”。
与表型图一样,我们首先基于PCA空间中的欧氏距离构建KNN图,并基于任意两个细胞在其局部邻域的共享重叠(Jaccard相似性)来细化边缘权值。这个步骤是使用FindNeighbors()函数执行的,并将之前定义的数据集维度(前10个pc)作为输入。
为了聚类细胞,我们下一步应用模块化优化技术,如Louvain算法(默认)或SLM [SLM, Blondel等人,Journal of Statistical Mechanics],迭代地将细胞分组在一起,以优化标准模块化函数为目标。FindClusters()函数实现了这个过程,并包含一个用于设置下游集群的“粒度”的解析参数,值的增加会导致集群数量的增加。我们发现,对于3K左右的单细胞数据集,在0.4-1.2之间设置该参数通常会返回良好的结果。对于较大的数据集,最佳分辨率通常会增加。可以使用Idents()函数找到集群。
pbmc <- FindNeighbors(pbmc, dims = 1:10)
## Computing nearest neighbor graph
## Computing SNN
pbmc <- FindClusters(pbmc, resolution = 0.5)
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 2638
## Number of edges: 95965
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.8723
## Number of communities: 9
## Elapsed time: 0 seconds
# Look at cluster IDs of the first 5 cells
head(Idents(pbmc), 5)
## AAACATACAACCAC-1 AAACATTGAGCTAC-1 AAACATTGATCAGC-1 AAACCGTGCTTCCG-1
## 2 3 2 1
## AAACCGTGTATGCG-1
## 6
## Levels: 0 1 2 3 4 5 6 7 8
非线性降维(UMAP/tSNE)
Seurat提供了几种非线性降维技术,如tSNE和UMAP,来可视化和探索这些数据集。这些算法的目标是学习数据的底层流形,以便在低维空间中放置相似的细胞。上面确定的基于图的集群中的单元应该在这些降维图上共定位。作为UMAP和tSNE的输入,我们建议使用相同的pc作为聚类分析的输入。
# If you haven't installed UMAP, you can do so via
# reticulate::py_install(packages = 'umap-learn')
pbmc <- RunUMAP(pbmc, dims = 1:10)
# note that you can set `label = TRUE` or use the LabelClusters function to
# help label individual clusters
DimPlot(pbmc, reduction = "umap")
此时,您可以保存对象,这样就可以轻松地将其加载回去,而无需重新运行上面执行的计算密集型步骤,或者轻松地与协作者共享。
saveRDS(pbmc, file = "./filtered_gene_bc_matrices/pbmc_tutorial.rds")
差异表达基因(聚类生物标志物)
Seurat可以帮助您找到通过差异表达定义集群的标记。默认情况下,与所有其他单元相比,它识别单个集群(在ident1中指定)的正标记和负标记。FindAllMarkers()为所有集群自动执行这个过程,但是您也可以对集群组进行对比测试,或者对所有单元进行测试。
pct参数要求在两组单元格中以最小百分比检测一个特性,以及阈值。测试论证需要一个特征在两组之间被差异表达(平均)。您可以将这两个值都设置为0,但是需要大幅增加时间—因为这将测试大量不太可能具有高度歧视性的特性。作为加速这些计算的另一个选项,可以设置max.cells.per.ident。这将向下采样每个标识类,使其单元格不多于设置的单元格。虽然通常会有功率的损失,但速度的提高可以是显著的,最高度不同的表现特征可能仍然会上升到顶部。
# find all markers of cluster 2
cluster2.markers <- FindMarkers(pbmc, ident.1 = 2, min.pct = 0.25)
head(cluster2.markers, n = 5)
## p_val avg_log2FC pct.1 pct.2 p_val_adj
## IL32 2.593535e-91 1.2154360 0.949 0.466 3.556774e-87
## LTB 7.994465e-87 1.2828597 0.981 0.644 1.096361e-82
## CD3D 3.922451e-70 0.9359210 0.922 0.433 5.379250e-66
## IL7R 1.130870e-66 1.1776027 0.748 0.327 1.550876e-62
## LDHB 4.082189e-65 0.8837324 0.953 0.614 5.598314e-61
# find all markers distinguishing cluster 5 from clusters 0 and 3
cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25)
head(cluster5.markers, n = 5)
## p_val avg_log2FC pct.1 pct.2 p_val_adj
## FCGR3A 2.150929e-209 4.267579 0.975 0.039 2.949784e-205
## IFITM3 6.103366e-199 3.877105 0.975 0.048 8.370156e-195
## CFD 8.891428e-198 3.411039 0.938 0.037 1.219370e-193
## CD68 2.374425e-194 3.014535 0.926 0.035 3.256286e-190
## RP11-290F20.3 9.308287e-191 2.722684 0.840 0.016 1.276538e-186
# find markers for every cluster compared to all remaining cells, report only
# the positive ones
pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
library(dplyr)
pbmc.markers %>%
group_by(cluster) %>%
slice_max(n = 2, order_by = avg_log2FC)
## # A tibble: 18 × 7
## # Groups: cluster [9]
## p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
##
## 1 1.17e- 83 1.33 0.435 0.108 1.60e- 79 0 CCR7
## 2 1.74e-109 1.07 0.897 0.593 2.39e-105 0 LDHB
## 3 0 5.57 0.996 0.215 0 1 S100A9
## 4 0 5.48 0.975 0.121 0 1 S100A8
## 5 7.99e- 87 1.28 0.981 0.644 1.10e- 82 2 LTB
## 6 2.61e- 59 1.24 0.424 0.111 3.58e- 55 2 AQP3
## 7 0 4.31 0.936 0.041 0 3 CD79A
## 8 9.48e-271 3.59 0.622 0.022 1.30e-266 3 TCL1A
## 9 4.93e-169 3.01 0.595 0.056 6.76e-165 4 GZMK
## 10 1.17e-178 2.97 0.957 0.241 1.60e-174 4 CCL5
## 11 3.51e-184 3.31 0.975 0.134 4.82e-180 5 FCGR3A
## 12 2.03e-125 3.09 1 0.315 2.78e-121 5 LST1
## 13 6.82e-175 4.92 0.958 0.135 9.36e-171 6 GNLY
## 14 1.05e-265 4.89 0.986 0.071 1.44e-261 6 GZMB
## 15 1.48e-220 3.87 0.812 0.011 2.03e-216 7 FCER1A
## 16 1.67e- 21 2.87 1 0.513 2.28e- 17 7 HLA-DPB1
## 17 3.68e-110 8.58 1 0.024 5.05e-106 8 PPBP
## 18 7.73e-200 7.24 1 0.01 1.06e-195 8 PF4
Seurat有几个测试的差异表达,可以设置与测试。使用参数(详见我们的DE插图)。例如,ROC测试返回任何单个标记的“分类能力”(范围从0 -随机,到1 -完美)。
cluster0.markers <- FindMarkers(pbmc, ident.1 = 0, logfc.threshold = 0.25, test.use = "roc",
only.pos = TRUE)
我们包括几个用于可视化标记表达式的工具。VlnPlot()(显示跨集群的表达式概率分布)和FeaturePlot()(在tSNE或PCA图上可视化特征表达式)是我们最常用的可视化。我们还建议探索RidgePlot()、CellScatter()和DotPlot()作为查看数据集的附加方法。
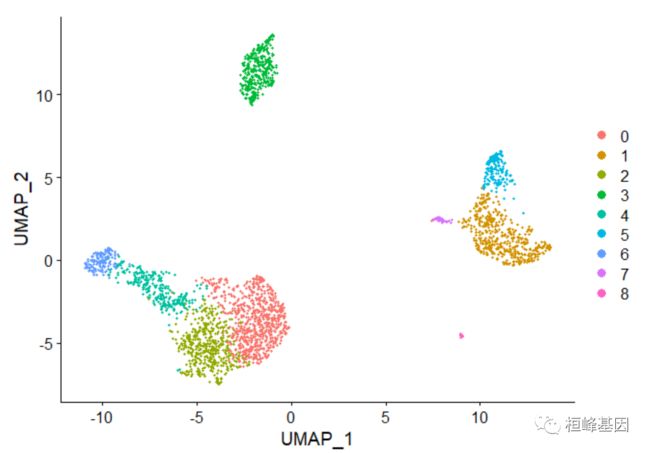
VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
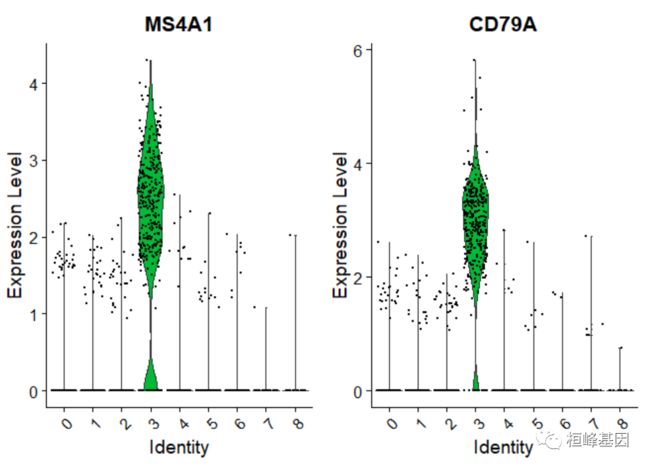
您也可以绘制原始计数
# you can plot raw counts as well
VlnPlot(pbmc, features = c("NKG7", "PF4"), slot = "counts", log = TRUE)
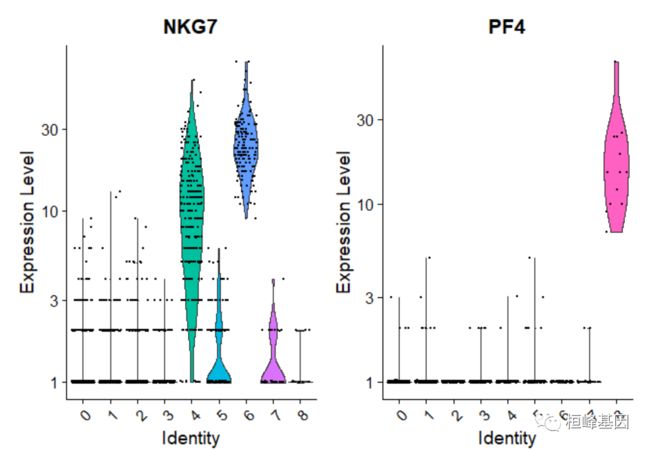
绘制每个基因的聚类图
FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A",
"LYZ", "PPBP", "CD8A"))
DoHeatmap()生成给定单元格和特征的表达式热图。在本例中,我们绘制每个集群的前20个标记(如果小于20,则绘制所有标记)。
pbmc.markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC) -> top10
DoHeatmap(pbmc, features = top10$gene) + NoLegend()

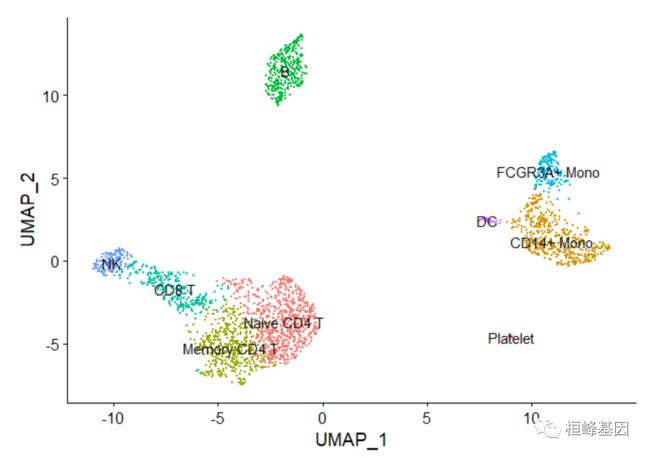
为聚类分配细胞类型
幸运的是,在这个数据集的情况下,我们可以使用规范的标记来轻松匹配已知细胞类型的无偏聚类:
Cluster ID Markers Cell Type
0 IL7R, CCR7 Naive CD4+ T
1 CD14, LYZ CD14+ Mono
2 IL7R, S100A4 Memory CD4+
3 MS4A1 B
4 CD8A CD8+ T
5 FCGR3A, MS4A7 FCGR3A+ Mono
6 GNLY, NKG7 NK
7 FCER1A, CST3 DC
8 PPBP Platelet
绘制聚类图
new.cluster.ids <- c("Naive CD4 T", "CD14+ Mono", "Memory CD4 T", "B", "CD8 T", "FCGR3A+ Mono",
"NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
saveRDS(pbmc, file = "./filtered_gene_bc_matrices/pbmc3k_final.rds")
所有的可视化分析就基本完成了,学会了单细胞分析就非常简单了,目前测序的费用也在降低,单细胞系列可算是目前的测序神器,有这方面需求的老师,联系桓峰基因,提供最高端的科研服务!
桓峰基因,铸造成功的您!
未来桓峰基因公众号将不间断的推出单细胞系列生信分析教程,
敬请期待!!
有想进生信交流群的老师可以扫最后一个二维码加微信,备注“单位+姓名+目的”,有些想发广告的就免打扰吧,还得费力气把你踢出去!
References:
- Hafemeister, C., Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296 (2019). https://doi.org/10.1186/s13059-019-1874