R语言TCGA数据下载及处理biolinks包的学习与使用(一)数据下载
biolinks包是一款非常方便的R包,可以直接处理很多TCGA数据

首先安装本包和其中一些相关的包,下载数据至少需要前两个包,后面的是生存分析需要,可以先不下载。如果不好下载建议上梯子。
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
library(BiocManager)
BiocManager::install("TCGAbiolinks")
library(TCGAbiolinks)
install.packages("survminer")
library(survminer)
BiocManager::install("SummarizedExperiment")
library(SummarizedExperiment)biolink包的核心函数在于:GDCquery,有11个参数:
1.project;
2.data.category;
3.data.type;
4.workflow.type;
5.legacy = FALSE;
6.access;
7.platform;
8.file.type;
9.barcode;
10.experimental.strategy;
11.sample.type
gbbTCGAbiolinks: Searching GDC database 本包官方教程
1.project:
也就是要下载的肿瘤的类型,使用代码查看肿瘤类型,缩写要自己去查是什么,本文用的是最常见的BRCA也就是乳腺癌了
TCGAbiolinks:::getGDCprojects()$project_id #查看有哪些癌症
[1] "TCGA-BRCA" "GENIE-MSK"
[3] "GENIE-VICC" "GENIE-UHN"
[5] "CPTAC-2" "CMI-ASC"
[7] "BEATAML1.0-COHORT" "CGCI-BLGSP"
[9] "BEATAML1.0-CRENOLANIB" "CMI-MPC"
[11] "CMI-MBC" "GENIE-GRCC"
[13] "GENIE-MDA" "GENIE-JHU"
[15] "GENIE-NKI" "FM-AD"
[17] "VAREPOP-APOLLO" "WCDT-MCRPC"
[19] "GENIE-DFCI" "TARGET-ALL-P3"
[21] "TARGET-ALL-P2" "OHSU-CNL"
[23] "TARGET-ALL-P1" "MMRF-COMMPASS"
[25] "TARGET-CCSK" "ORGANOID-PANCREATIC"
[27] "NCICCR-DLBCL" "TARGET-NBL"
[29] "TARGET-OS" "TARGET-RT"
[31] "TARGET-WT" "TCGA-LAML"
[33] "CGCI-HTMCP-CC" "TARGET-AML"
[35] "HCMI-CMDC" "TCGA-DLBC"
[37] "TCGA-CHOL" "CTSP-DLBCL1"
[39] "TRIO-CRU" "TCGA-MESO"
[41] "TCGA-ACC" "TCGA-UCS"
[43] "TCGA-KICH" "TCGA-PCPG"
[45] "TCGA-ESCA" "TCGA-THYM"
[47] "TCGA-TGCT" "TCGA-UVM"
[49] "TCGA-CESC" "TCGA-BLCA"
[51] "TCGA-PAAD" "TCGA-LIHC"
[53] "TCGA-SKCM" "TCGA-UCEC"
[55] "TCGA-PRAD" "REBC-THYR"
[57] "TCGA-THCA" "TCGA-OV"
[59] "TCGA-LGG" "TCGA-SARC"
[61] "CPTAC-3" "TCGA-COAD"
[63] "TCGA-READ" "TCGA-KIRP"
[65] "TCGA-GBM" "TCGA-STAD"
[67] "TCGA-LUAD" "TCGA-KIRC"
[69] "TCGA-LUSC" "TCGA-HNSC" 要下载乳腺癌,即
project="TCGA-BRCA"2.data.category:
数据类别,包括转录图谱、甲基化数据、临床数据等多种数据类型
TCGAbiolinks:::getProjectSummary("TCGA-BRCA")#查看有哪些分类数据
$file_count
[1] 34686
$data_categories
file_count case_count data_category
1 8648 1044 Simple Nucleotide Variation
2 919 881 Proteome Profiling
3 1183 1098 Clinical
4 5316 1098 Biospecimen
5 6080 1097 Transcriptome Profiling
6 6627 1098 Copy Number Variation
7 4679 1098 Sequencing Reads
8 1234 1095 DNA Methylation
$case_count
[1] 1098
$file_size
[1] 5.707689e+13case_count为病人数,file_count为对应的文件数,我们下载基因表达数据
data.category = "Gene expression"3.data.type
数据类型,常见的有以下三个
#下载rna-seq的counts数据
data.type = "Gene Expression Quantification"
#下载miRNA数据
data.type = "miRNA Expression Quantification"
#下载Copy Number Variation数据
data.type = "Copy Number Segment"我们下载基因表达量化数据
data.type = "Gene expression quantification"4.workflow.type
工作流程,有以下三个供选择
-
HTSeq - FPKM-UQ:FPKM上四分位数标准化值
-
HTSeq - FPKM:FPKM值/表达量值
-
HTSeq - Counts:原始count数
概念:
计算:


要求出FPKM值,需要获得三个参数。
cDNA Fragments:可以理解为单个基因比对到基因组上的reads数,在测序数据里就是count值。HTseq处理后可以直接得到结果。
Mapped Fragments:指每个样品中所有基因比对到基因组上的reads数。也就是用求和函数sum()将单一样品的count全部加起来。
Transcript Length:也就是exon length,是指reads比对到基因外显子上的长度。这个需要找到参考基因组才能获得数据。
区别:简单的讲,Counts是数据后台没有处理的原始表达量,而FPKM和FPKM-UQ是两种数据处理方法,也就是说,如果下载Counts数据,是表达量数据,如果下载FPKM数据,那么要注意这些数据是经过处理的。下载数据后,在数据分析时,用的方法也是不同的,Counts数据一般使用edgeR包或DESeq包,对数据做分析;如果下载FPKM数据,就不能使用edgeR包,只能只用DESeq包进行处理。在使用edgeR包做Counts数据处理时,是需要对数据进行normalize的,所以我们在下载数据时,下载counts是比较常用的。
workflow.type=“HTSeq - Counts”5.legacy
这个参数主要是因为TCGA数据有两个入口可以下载,GDC Legacy Archive 和 GDC Data Portal,区别主要是注释参考基因组版本不同分别是:GDC Legacy Archive(hg19)和GDC Data Portal(hg38)。参数默认为FALSE,下载GDC Data Portal(hg38)。下载转录组层面的数据使用hg38,下载DNA层面的数据使用hg19,因为比如做SNP分析的时候很多数据库没有hg38版本的数据,都是hg19的。
至于hg19和hg38的区别,中文网上没有很好的答案,我自己搜索了以下,大概就是在做肿瘤单核苷酸变异(single nucleotid variant,SNV)的时候,是需要参考正常人的whole genome sequencing(WGS)结果,然后将短的测序reads匹配到参考基因组。目前WGS已经有很多代的结果,越来越先进,所以hg38是比19要更加准确。但有的时候需要在先前的工作基础上继续工作;或者需要和先前的工作对比,那就需要用回hg19,二者有工具可以切换。
6.access
数据开放和不开放,有两个参数:controlled, open。
我们这里使用:access=“open”
7.platform
平台有很多,可以自己去官网看需要什么平台,我们用Illumina
platform = "Illumina HiSeq"8.barcode
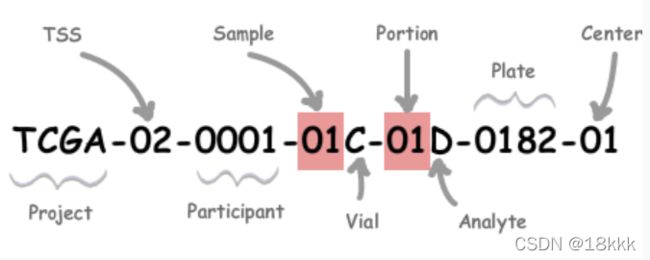
这个是说可以指定只下载某些样本,我们为了数据处理快,就定义一些样本来下载,默认会下载所有样本
barcode的命名还是有一些规律的,但个人感觉不太重要哈
listSamples <- c("TCGA-E9-A1NG-11A-52R-A14M-07","TCGA-BH-A1FC-11A-32R-A13Q-07",
"TCGA-A7-A13G-11A-51R-A13Q-07","TCGA-BH-A0DK-11A-13R-A089-07",
"TCGA-E9-A1RH-11A-34R-A169-07","TCGA-BH-A0AU-01A-11R-A12P-07",
"TCGA-C8-A1HJ-01A-11R-A13Q-07","TCGA-A7-A13D-01A-13R-A12P-07",
"TCGA-A2-A0CV-01A-31R-A115-07","TCGA-AQ-A0Y5-01A-11R-A14M-07")barcode = listSamples
9.file.type
下载数据hg19版本数据的时候使用,可以参考官网说明。这里设置为
file.type = "results"如果你前面的legacy设置为F(默认),那么这里你下载hg38的时候就不需要设置这个选项了
10.experimental.strategy
两个下载入口参数选择,可以默认

11.sample.type
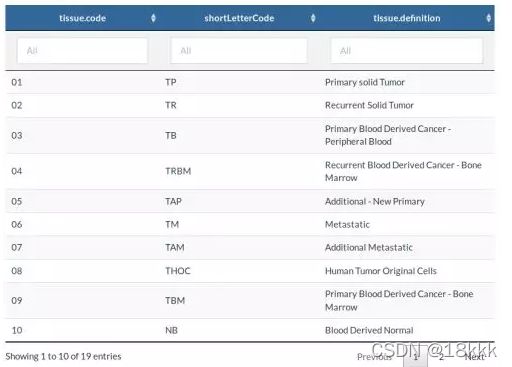
可以对样本类型进行过滤下载。要下载所有样本类型数据,不设置。部分值选择如下(全部可以查看官网):如sample.type = "Recurrent Solid Tumor"大概就是说下载什么类型的肿瘤。
开始下载数据
listSamples <- c("TCGA-E9-A1NG-11A-52R-A14M-07","TCGA-BH-A1FC-11A-32R-A13Q-07",
"TCGA-A7-A13G-11A-51R-A13Q-07","TCGA-BH-A0DK-11A-13R-A089-07",
"TCGA-E9-A1RH-11A-34R-A169-07","TCGA-BH-A0AU-01A-11R-A12P-07",
"TCGA-C8-A1HJ-01A-11R-A13Q-07","TCGA-A7-A13D-01A-13R-A12P-07",
"TCGA-A2-A0CV-01A-31R-A115-07","TCGA-AQ-A0Y5-01A-11R-A14M-07")
query <- GDCquery(project = "TCGA-BRCA",
data.category = "Gene expression",
data.type = "Gene expression quantification",
experimental.strategy = "RNA-Seq",
platform = "Illumina HiSeq",
file.type = "results",
barcode = listSamples,
legacy = TRUE,
access="open"
workflow.type = "HTSeq - Counts")
GDCdownload(query)下载成功以后,在工作环境的目录下就会多一个文件夹,里面是TCGA的数据

接下来本包还可以进行后续的下游分析,有时间再更