Chapter22: Artificial Intelligence in Drug Safety and Metabolism
reading notes of《Artificial Intelligence in Drug Design》
文章目录
- 1.Introduction
- 1.1.History of QSAR
- 1.2.History of Toxicology
- 1.3.History of Computational Toxicology
- 2.Method
-
- 2.1.A Model and its Usefulness
- 2.2.Model Accuracy
- 2.3.Applicability Domain
- 2.4.The Importance of Similarity
- 2.5.Predicting Pharmacokinetics
- 3.Applications of Computational Toxicology Models
-
- 3.1.Cardoac Toxicology
- 3.2.Hepatic Toxicology
- 3.3.Genetic Toxicology
- 3.4.In Vivo Pharmacokinetics
- 3.5.Digital Pathology
- 4.Future Outlook
1.Introduction
- The overarching concept of machine learning has been in common use in small molecule pharmaceuticals for over 30 years and forms a strong part of the medicinal chemistry drug design process in almost all drug companies.
1.1.History of QSAR
- QSAR (quantitative structure activity relationship) is developed by the work of Hansch based initially on whole molecule Hammett physicochemical parameters representing steric, electrostatic and hydrophobic properties. These were used to predict very similar molecules in early QSARs.
- Free and Wilson evolved this idea to apply the concept of smaller molecular fragmental contributions summed up additively to give a model the whole molecules overall property.
- Molecular descriptors have evolved from these concepts into ever larger more complex mathematical objects such as molecular graphs, which is outside the scope of this chapter but have been recently reviewed by David.
1.2.History of Toxicology
- Since the late 1980’s the Organization for the Economic Co-operation and Development (OECD) and the International Conference on Harmonization (ICH) have introduced widely accepted global guidance for the toxicity testing of pharmaceutical products for regulatory purposes.
1.3.History of Computational Toxicology
- Compared to other applications of AI, it may be that the safety pharmacology endpoints being measured are less precise and more difficult to describe in terms of molecular descriptors than simpler experimental endpoints.
- The measurements often come with high experimental variance and high cost associated with phenotypic and in vivo experiments which can involve many complex biological pathways and processes.
- The data from drug discovery programs is often sparse since not all projects use the same screening cascades or deal with the same issues and there are many fewer compounds at the stage in the pipeline where safety becomes most important.
- Finding datasets of sufficient size and quality is often still a challenge for computational toxicology.
2.Method
2.1.A Model and its Usefulness
- A model is successful if it can be used to guides useful decisions.
- A model builder must understand how accurate does the model really need to be given the use case and how accurate can it be given the data on which it is built.
- Another consideration is can it be used to prioritize resources and give better than human intuition performance.
- When an assay is high throughput but perhaps fast and cheap, it potentially has a larger error than a more specific, costly, and more relevant assay. It is not uncommon that models built on this high-throughput data will be of inherently poorer quality.
2.2.Model Accuracy
- Different error include
- Data errors inherent in the experimental error of complex safety assays.
- The inherent error due inaccuracy or imprecision of chemical descriptors in describing the properties of a molecule.
- Statistical analysis errors associated with different mathematical modeling techniques like overfitting or unnecessarily complex models.
2.3.Applicability Domain
- There are some principles about Applicability Domain (AD):
- AD study assures the reliability of predictions for new molecules. Approaches such as the TARDIS principle, which stands for transparency, applicability, reliability, decidability, interpretability, and support, are most useful.
- Can the prediction from the test compound be reliable because of high similarity to compounds in the test set using probability, geometric, or distance-based methods?
- Was the choice of training set examples specific to defining a given AD?
- Reliability or error in the data values (measured by experimentally derived standard deviation) sets the limits within which a QSAR results can be meaningfully interpreted.
- Understanding the uncertainty of model and measurement determines whether there is any value in using the model to make decisions based on predictions.
- Estimation of AD is dependent on many factors such as algorithm, data distribution, endpoint value, and descriptor set.
- Frequently time based train–test splits can come close to approaching the actual real world use case for these models even if they are less flattering in measures of accuracy than typical random splits.
2.4.The Importance of Similarity
- Three main similarity approaches are used in safety prediction:
- Methods of grouping by property or structure similarity like read across (RAX) and categorization.
- QSAR as previously discussed.
- Expert systems rule based or decision trees.
- The comparison of descriptor sets has been effectively summarized recently by Todeschini and many of those in common use perform equally well.
2.5.Predicting Pharmacokinetics
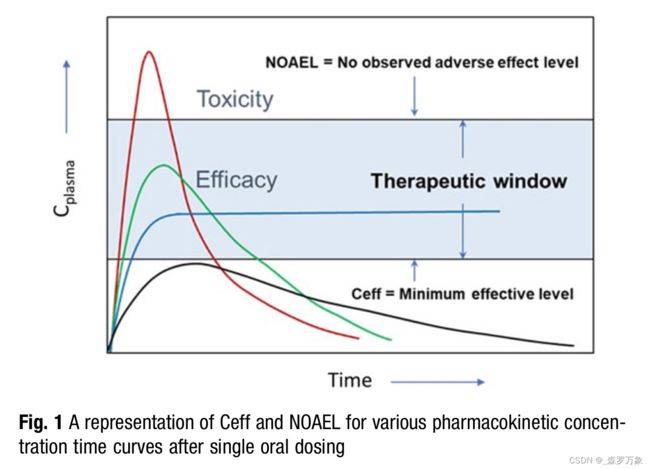
- Many safety outcomes are driven by maximum plasma concentration (Cmax) achieved, whereas many efficacy measurements are driven by multiples of minimum plasma concentration (Cmin) or area under the time-concentration curve (AUC).
- No observed adverse effect level (NOAEL) concentration defines the upper bound of safe concentration and is determines in in vivo safety studies.
- The efficacious concentration (Ceff) is determined in PD efficacy studies. Figure 1. The safety margin for a drug is simply [NOAEL]/[Ceff], ideally a positive integer greater than 10 to allow for any population variance in the clinical setting.
3.Applications of Computational Toxicology Models
3.1.Cardoac Toxicology
- The most well-known interaction is the binding of molecules to the voltage gated ion-channel Kv11.1 (hERG) which regulated cardiac action potential. hERG inhibition is linked with long QT Syndrome and is associated with torsade’s de pointes, a potentially fatal condition.
- Cai reported a multitask deep neural network built on 7889 datapoints from public sources which accurately predicted in vitro hERG activity.
- Zhang has also reported effective deep learning models of hERG which performed better than by other methods. These models were built on 697 molecules from the literature.
- Adedinsenwo and Hannun recently showed that using a convolutional neural network (the AI-ECG algorithm) to diagnose left ventricular systolic dysfunction performed better than standard of care which was blood testing for N-terminal pro-B–type natriuretic peptide.
3.2.Hepatic Toxicology
- Drug induced liver injury (DILI) is the leading cause for drug withdra- wals in the USA.
- Williams recently showed that using many preclinical in vitro measures of hepatotoxicity as features in an AI model using Bayesian mathematics they are able to produce an accurate predictor of clinical DILI. Another feature of this model is that due to the probabilistic nature of the method it is able to represent the uncertainty in the prediction in the output graphically which enables human interpretation when choosing which compounds to progress (Fig. 2).
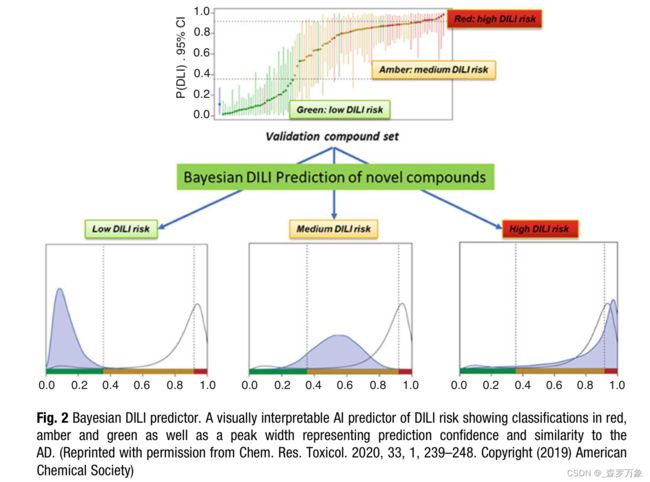
- Semenova recently reported a similar approach using a Bayesian Neural Network (BNN) to model the DILI endpoint in what is perhaps the first application of a BNN applied to toxicity prediction.
- Minerali used the same AstraZeneca-Pfizer data set and compared various common AI techniques to evaluate relative model performance.
- Hammann constructed a DILI prediction model with an accuracy of up to 89% using AI methods based on a large set of DILI annotated drugs using physicochemical descriptors.
3.3.Genetic Toxicology
- The most cost effective computational toxicology models, although not the most sophisticated, are perhaps the structure alerts.
3.4.In Vivo Pharmacokinetics
- One difficulty for AI in this area is that datasets are not large in terms of number of molecules measured relative to other drug discovery endpoints. With the application of deep learning methods comes 2 new possibilities not offered by most older AI techniques which are multitask models and transfer learning. These techniques offer opportunities to expand the AD outside of the original data set using other related data.
- Lowe has used several AI techniques including ANN, support vector machines (SVM) with the extension for regression and kappa nearest neighbor (KNN). These combined with 2D and 3D descriptor sets were used to evaluate models of rat and human in vitro PK parameters such as liver microsomal intrinsic clearance (HLM) and fraction unbound ( fu) for 400–600 sampled compounds from the literature.
- In more recent studies, Wang built 2D and 3D QSAR models using several AI techniques for 1352 drugs from a large public data set for which there was human intravenous PK data. In this work clearance model accuracy measures (R2 and RMSE) were better than for those obtained before for in vivo clearance modeling.
- Schneckener used a data set of 1882 high quality in vivo rat PK experiments from the Bayer collection to build QSAR models of intravenous and oral exposure and afterward calculate oral bioavailability.
- Feinberg has built models of 31 physicochemical and DMPK assays in the Merck collection using more complex multitask molecular graph convolutions from a graph convolutional neural network (GCNN) as descriptors and achieving “unprecedented accuracy in prediction of ADMET prop- erties” in a large and well validated study which compared single task GCNN and random forest methods.
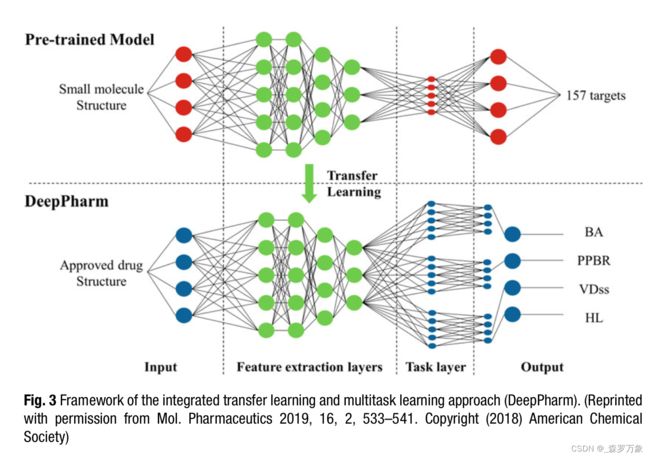
- Ye was able to build AI models of 4 human PK parameters from a data set of 1104 Federal Drug Administration approved small molecule drugs named DeepPharm. Using an ANN model pretrained on over 30 million publicly available bioactivity datapoints they then used transfer learning to a multitask ANN to enhance the model’s generalization across the ADME endpoints (Fig. 3).
3.5.Digital Pathology
- In the area of digital pathology great improvements are being made in speed and accuracy by using AI based image recognition techniques applied to newly developed digital whole slide microscope (WSM) images.
- Tokarz developed a computer-assisted image analysis algorithm to quantify the common features of rodent progressive cardiomyopathy (PCM) in images of rat heart histologic sections.
4.Future Outlook
- In some of these lower-throughput endpoints, we might hope to see more cross industry data or model sharing as seen in such recent consortia as the European machine learning ledger orches- tration for drug discovery (MELLODDY) project. Through sharing the data of 10 pharmaceutical companies the consortium has access to >10 million small molecule structures and >1 billion associated high-throughput assay values. MELLODDY aims to build better AI models, using methods like massive multitask learning using a centralized AI platform. This group will model on the largest achievable scale without exposing proprietary information between members.