用Gromacs重复文献计算TIP4P介电常数谱
重复文献Classical molecular dynamics simulation of microwave heating of liquids中对于TIP4P水介电常数的计算
很多人刚开始做gromacs都是从重复文献开始,但是gromacs输入文件输出文件参数复杂,对于新手很不友好,需要弄清处理各个文件的意义,才能正确的使用gromacs,所以在此具体地展示如何根据文献中的参数对Gromacs的不同文件进行设置。这篇文献需要在外网获得,我直接放在资源里了,可以免费获得。
文件夹在桌面packmol里
明确重复对象
首先找到文献中关于TIP4P中的结果,一个是频率响应的介电常数谱,另一个是静止介电系数。
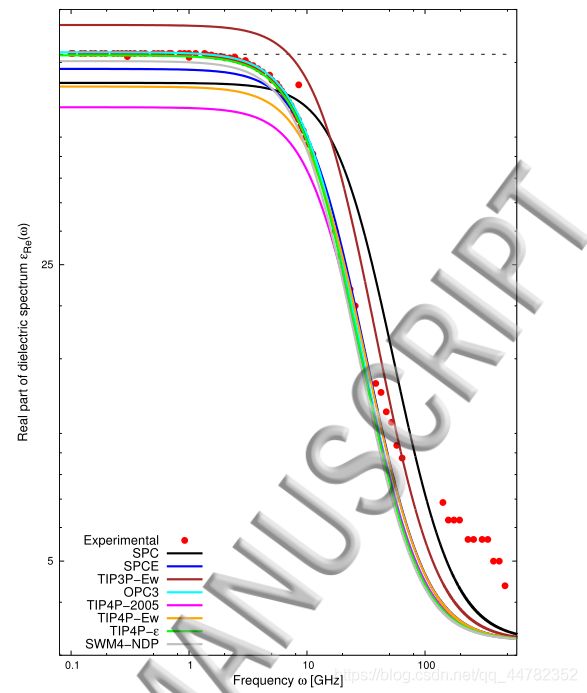
We obtained the following average values of the static dielectric constant of water: 67.1
± 2.2 (SPC), 72.4 ± 2.1 (SPCE), 91.9 ± 2.0 (TIP3P-Ew), 79.3 ± 1.9 (OPC3), 58.8 ± 1.6
(TIP4P-2005), 65.8 ± 2.1 (TIP4P-Ew), 77.9 ± 2.0 (TIP4P–ϵ), 75.5 ± 2.0 (SWM4-NDP).
Our results agree well with the following computational studies: 65.0 (SPC18), 71.0 (SPCE5),89.0 (TIP3P-Ew6), 78.4 (OPC38), 60.0 (TIP4P-20057), 63.4 (TIP4P-Ew7), 78.3 (TIP4P-ϵ10),and 79.0 (SWM4-NDP9).
从这段话中找到TIP4P-2005的信息,我们知道算出来的结果应该是58.8左右这个数附近。
使用Gromacs计算
因为gromacs自带的文件夹中就已经拥有tip4p.gro这个文件了,这是tip4p水分子的一个仿真盒子,我们可以通过这个文件得到文献中的仿真盒子。
我们看 COMPUTATIONAL DETAILS这一节,下面有说明其中的参数设置。
For each empirical force field four different sets of classical MD simulations are carried
out. All simulations employed 3000 water molecules in a cubic simulation box. This sample
size was decided on the basis of the size-dependence of the different dielectric properties
obtained from exploratory simulations. A time step of 1.0 fs was used for all simulations,
with periodic boundary conditions (PBC) applied in all directions to mimic 3D bulk samples.
Long-range Coulombic interactions were evaluated using the particle-particle/particle-mesh
(PPPM) solver3, using a precision factor of 0.0001 and a real space cut-off of 10.0˚A. The
short-range interaction cut-off was set also to 10.0˚A. For all simulations bonds involving hydrogen atoms were constrained during dynamics using the Shake algorithm11to allow for
using a time step of 1.0 fs. All simulations were conducted at 298.0 K and 1 atmosphere of
pressure. In the case of the SWM4-NDP polarizable force field the temperature of Drude
particles was kept as low as 1.0 K in order to maintain the induced dipoles near the self-
consistent induction regime9.
这里我先翻译一下
对于每个经验力场,分别进行了四组不同的经典MD模拟。所有的模拟都使用了一个立方体模拟盒中的3000个水分子。这个样本大小是基于从探索性模拟中获得的不同介电特性的尺寸依赖关系来确定的。所有的模拟都采用1.0fs的时间步长,并在所有方向上应用周期性边界条件(PBC)来模拟3D整体样品。使用粒子-粒子/粒子-网格(PPPM)解算器3,使用0.0001的精度因子和10.0˚A的实际空间截止值,评估了长程库仑相互作用。对于涉及的所有模拟键,短程相互作用截止值也被设置为10.0˚A。对于涉及以下内容的所有模拟键合氢原子在动力学过程中使用Shake算法11进行约束,以允许使用1.0fs的时间步长。所有的模拟都是在298.0 K和1个大气压下进行的。在SWM4-NDP极化力场的情况下,德鲁德粒子的温度保持在1.0K以下,以便将诱导偶极子保持在自洽诱导方案9附近。
这段话中首先告诉我们,他用有3000个TIP4P,但是没有给出水分子仿真盒子的大小,经过我的测试,使用盒子的尺寸为4.11 4.5 5的时候刚好有3000个水分子,所以输入以下代码
gmx solvate -cs tip4p.gro -o conf.gro -box 4.11 4.5 4.9 -p topol.top
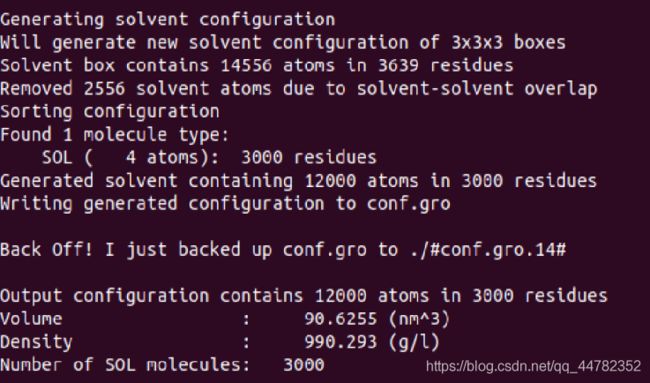
这样就生成了用于后面计算的top和gro文件。
然后继续阅读上面的文字,有用的信息是dt = 1fs ,还有10.0˚A的实际空间截止值,还有约束算法使用shake,温度是298K。这些都需要在.mdp文件中修改。
mdp文件
打开nvt.mdp文件(window系统中右键编辑)
title = Protein-ligand complex NVT equilibration
define = -DPOSRES ; position restrain the protein and ligand
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 100000 ; 1 * 100 = 100 ps
dt = 0.001 ; 1 fs
; Output control
nstxout = 1000 ; save coordinates every 1.0 ps
nstvout = 1000 ; save velocities every 1.0 ps
nstenergy = 1000 ; save energies every 1.0 ps
nstlog = 1000 ; update log file every 1.0 ps
energygrps = system
; Bond parameters
continuation = no ; first dynamics run
constraint_algorithm = shake ; holonomic constraints
constraints = all-bonds ; all bonds (even heavy atom-H bonds) constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighborsearching
cutoff-scheme = Verlet
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet
rcoulomb = 1 ; short-range electrostatic cutoff (in nm)
rvdw = 1 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = system ; two coupling groups - more accurate
tau_t = 0.1 ; time constant, in ps
ref_t = 298 ; reference temperature, one for each group, in K
; Pressure coupling
pcoupl = no ; no pressure coupling in NVT
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction
DispCorr = EnerPres ; account for cut-off vdW scheme
; Velocity generation
gen_vel = yes ; assign velocities from Maxwell distribution
gen_temp = 298 ; temperature for Maxwell distribution
gen_seed = -1 ; generate a random seed
这里比较重要要改的是dt还有step,dt和step配合达到总的100ps,然后gen_temp要改到298,约束方式要改的shake,注意如果更改dt的话,一般文件记录的频率也要对应更改。
然后更改npt.mdp中的
title = Protein-ligand complex NPT equilibration
define = -DPOSRES -DPOSRES_LIG ; position restrain the protein and ligand
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 1900000 ; 1.9ns
dt = 0.001 ; 1 fs
; Output control
nstxout = 1000 ; save coordinates every 1.0 ps
nstvout = 1000 ; save velocities every 1.0 ps
nstenergy = 1000 ; save energies every 1.0 ps
nstlog = 1000 ; update log file every 1.0 ps
energygrps =
; Bond parameters
continuation = yes ; first dynamics run
constraint_algorithm = shake ; holonomic constraints
constraints = all-bonds ; all bonds (even heavy atom-H bonds) constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighborsearching
cutoff-scheme = Verlet
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet
rcoulomb = 1.0 ; short-range electrostatic cutoff (in nm)
rvdw = 1.0 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = system ; two coupling groups - more accurate
tau_t = 0.1 ; time constant, in ps
ref_t = 298 ; reference temperature, one for each group, in K
; Pressure coupling
pcoupl = Parrinello-Rahman ; pressure coupling is on for NPT
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 2.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
refcoord_scaling = com
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction
DispCorr = EnerPres ; account for cut-off vdW scheme
; Velocity generation
gen_vel = no ; velocity generation off after NVT
更改的内容与nvt.mdp文件中的类似,rcoulomb和rvdw对应库仑力与范德华力作用,要按照文献中更改为1.0。
使用Gromacs按照其内容流程出图
文献中提到进行了100ps的nvt和npt过程后计算介电常数,所以按照流程制作即可。
能量最小化
gmx grompp -f minim.mdp -c conf.gro -p topol.top -o em.tpr
gmx mdrun -v -deffnm em
NVT预平衡
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr
gmx mdrun -v -deffnm nvt
NPT预平衡
gmx grompp -f npt.mdp -c nvt.gro -r nvt.gro -p topol.top -o npt.tpr
gmx mdrun -v -deffnm npt
计算偶极矩
gmx dipoles -corr total -c -f npt.trr -s npt.tpr
这步做完之后会产生静止介电常数
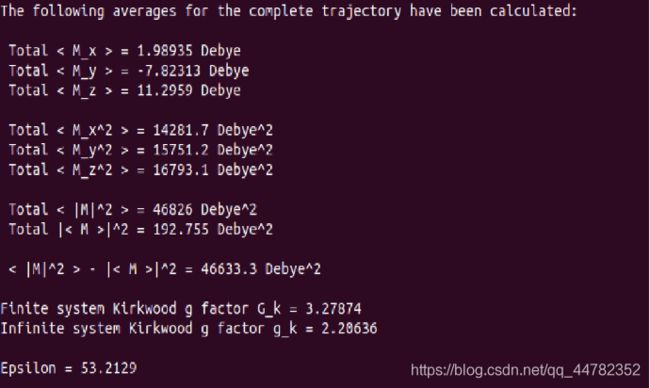
这个值与文献大概差了3,由于有一些参数在gromacs中无法完全仿照,这个结果还是可以接受的。
计算频率响应介电常数
gmx dielectric -f dipcorr.xvg -eps0 53.2129 -ffn aexp
这里的eps0是偶极距计算中的静止介电常数
显示图片
xmgrace epsw.xvg
用grace显示图片

gromacs自带的计算函数并不是很好,这里横坐标应该取log好一些。但是从这张图也可以看出来在0-300GHz这个区域与文献是一致的。这里我建议还是自己仿照他的程序自己编一个,这样看起来会美观很多。
至此,我们就完成TIP4P-2005水的文献重复过程,其他的分子可以以此作为参考。
总结
做科研一开始都是一个很难的过程,一开始都是要重复前人的工作,然后才能有自己更深的发现,希望看到这篇文章的你能坚持你的科研工作。因为即使程序bug再多,也有de完的一天。