DeepGS:Deep Representation Learning of Graphs and Sequences for DTA | CNN,GAT,BiGRU,Prot2Vec
DeepGS: Deep Representation Learning of Graphs and Sequences for Drug-Target Binding Affinity Prediction(DTA)
这是一篇预测药物和靶标结合亲和力的文章,发表在ECAI2020上。文章最大的创新点在于首次提出三通道,在此之前都是使用二通道预测。
ECAI 2020:European Conference on Artificial Intelligence 欧洲人工智能会议
一、Introduction
(1) 背景
近年来,随着深度学习模型在各个领域的成功应用与亲和力数据量的增加,深度学习技术被应用于DTA预测。而现存模型使用独热编码或只考虑分子的拓扑结构,而不考虑分子的局部化学背景。
(2)本文工作
本文提出了一个基于深度学习的 DTA预测模型DeepGS,将氨基酸序列和SMILES串 编码为分布式表示,而且同时考虑了药物的 分子结构和化学背景。作者将DeepGS与一些模型进行比较,实验结果证明了DeepGS的表现最好。
二、模型介绍
模型以 蛋白质序列、药物的分子结构 还有 药物的SMILES串作为输入,以 药物靶标结合亲和力 作为输出。
模型主要分为三个部分。使用CNN来学习蛋白质序列(提取特征),使用GAT和BiGRU来学习药物。
具体来说步骤如下:
step1:分别使用Prot2Vec和Smi2Vec将蛋白质和药物序列中的符号 编码成 分布式表示;并将序列转换为矩阵,其中每一行表示序列中的符号;
step2:从蛋白质矩阵、分子矩阵、分子结构图中提取特征。①对于蛋白质序列,我们考虑氨基酸的局部化学背景,通过CNN提取特征;②对于药物分子图,使用GAT提取药物的拓扑结构信息(后面详细讲);③使用BiGRU捕捉药物的局部化学背景。至此,我们获得了蛋白质的一个潜在表达和药物的两个潜在表达。
step3:DeepGS将三个潜在表示传递给神经网络以预测结合亲和力。
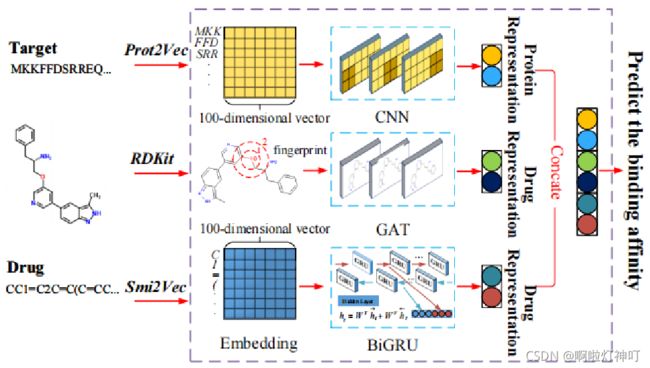
1.蛋白质的表示
(1)使用氨基酸序列表示靶蛋白。对于序列 L = { xi | i=1,2,…,l } ,采用N-gram算法将它分为三个一组的biological word :[x1,x2,x3],[x4,x5,x6],[x7,x8,x9]
(2)使用Prot2Vec将biological word 编码成d维向量,目标序列就转化成了一个矩阵,每一行都是一个生物单词的嵌入。
(3)将矩阵输入到CNN,提取蛋白质的化学环境。
(1)①20种氨基酸,每一种都有对应的字母缩写。
②序列中,xi 表示第i个氨基酸,l表示序列长度。
③N-gram设置:,N设为3(为了在训练可行性和词汇量之间进行权衡)。
N-gram介绍:N-Gram是一种基于 统计语言模型 的算法。它的基本思想是 将 文本里面的内容 按照字节 进行 大小为N的 滑动窗口操作,形成了长度是N的字节片段序列,每一个字节片段称为gram。
(2)Prot2Vec:用n维向量来表示蛋白质。对于每个biological word,从预训练的嵌入字典中查找每个单词的嵌入向量。
这个嵌入字典来自Swiss-Prot,Swiss-Prot是一个经过注释的 蛋白质序列 数据库,包含56万个手工注释的序列。
【Q】为什么使用Prot2Vec?【A】因为它可以捕获靶蛋白中的局部化学信息。
【Q】为什么采用N-garm模型?【A】因为单个氨基酸通常无意义。
2.药物的表示
(1)药物分子图
①使用RDKit工具包将SMILES串转化为分子图
②将 graph attention network (GAT)应用于分子图:
A. 计算每个原子的临边信息和分子指纹并拼成一个向量;
B. 通过传播相邻节点的信息来更新向量;
C. 通过聚合每个原子的向量来获得对分子的表示。

(1)RDKit是一个用于 化学信息学 的开源工具包,里面有很多将化学与机器学习联系起来的、非常实用的库。RDKit基于对化合物2D和3D分子操作,利用机器学习方法进行化合物描述符生成,fingerprint生成,化合物结构相似性计算,2D和3D分子展示等。基于PYTHON语言进行调取使用。
将SMILES串转化为图G:from rdkit import Chem,①将SMILES转化为RDKIT的mol对象,同时生成一个空的图;②提取原子的特征和化学键特征并加入图中。
(2)基于GAT的分子结构建模方法通过聚合 r-半径子图的表示 来提取药物的拓扑特征。
分子指纹:以r为半径的子图,使用WL算法(Weisfeiler-Lehman)来计算分子指纹。
a:给出两个标签 Label 的图
b:考虑节点邻域的标签,并对此排序。
c:对标签进行压缩映射
d:得到新标签
(2)药物化学背景
① 使用Smi2Vec将药物表示为100维的向量,组成矩阵;
② 使用BiGRU获得药物的潜在表示。

Smi2Vec算法介绍:
A. SMILES串被分成单个原子或字符 xj(长度为m);
B. 从预训练的字典中查找每个原子embedding来进行映射,若不在字典中就随机生成一个值;
C. 把embedding vector聚合起来组成一个矩阵(A) ,每一行表示表示一个原子预训练后的向量。
【Q】为什么不用one-hot?
【A】one-hot比较稀疏,而且不能表示语意之间的关系。Smi2Vec通过embedding将高维稀疏矩阵 转化为 低维稠密矩阵,而且具有相似语意的 映射在 空间的相似位置
BiGRU(双向BRU)采用固定大小的矩阵作为输入,而SMILES字符串的长度可能会有所不同,解决方案是:当训练BiGRU和CNN时,我们将输入序列的长度固定在某个值。如果输入序列的长度大于l,我们将裁剪输入序列;如果小于l,则在输入序列的末尾使用零填充。在“实验结果”部分的模型灵敏度部分会详细介绍。
> BiGRU介绍:
BiGRU结构:BiGRU由两个信息传递相反的GRU循环层构成,一种是按时间顺序传递信息,这有助于我们从以前的表述中学习,另一种是按时间逆序传递信息,这有助于我们从未来的表述中学习。这样每个时间步的输出节点,都包含了输入序列中当前时刻完整的过去和未来的上下文信息。
BiGRU作用:为了更好地理解上下文和消除歧义,我们需要从将来的时间步中学习表示。
(e,g)“He said,Teddy bear was on sale”,“He said,Teddy Roosevelt was a great president”。在以上两个句子中,当我们看到单词“Teddy”和前两个单词“He said”时,我们可能无法理解这个句子是指总统还是泰迪熊。因此,要解决这种不确定性,我们需要向前看。这就是双向RNNs的功能。
3.DTA预测
将前面三部分得到的信息送到全连接层来预测亲和力:
①激活函数:ReLU
②损失函数:均方误差 mean Square error
全连接层,充当分类器的作用。
激活函数,就是在 人工神经网络 的 神经元上 运行的函数,负责将神经元的输 入映射到输出端。
①ReLU:用于隐层神经元输出,公式为f(x) = max(0, x),是一个线性函数。
【Q】前面三部分信息怎么处理的?
【A】(1)对于CNN模块,用三个连续的2D卷积层;
(2)对于GAT模块,使用两个图注意力层来更新图中的节点向量(考虑它们的邻居节点);
(3)对于BiGRU模块,使用一个BiGRU层。
三、实验设置
(1)数据集
将数据集分成大小相同的6个子集,五份做训练集,一份做测试集:
①Davis数据集,包含68种药物、442种靶标、30056种药物-靶标相互作用。
②KIBA数据集,包含2111种药物、229种靶标、118254种药物-靶标相互作用。
(1)数据集介绍:
①Davis数据集是Davis 等人2011年发现的亲和力进行的实验的数据
②KIBA数据集是从ChEMBL和STITCH收集的,这确保了KIBA中的数据是经过实验验证的。KIBA数据集结合了各种生物活性类型,包括Kd,Ki和IC50,并消除不同生物活性类型之间的不一致,这大大降低了数据集中的偏差。
其中ChEMBL一个大型的、开放访问的 药物发现数据库,旨在收集 药物研究和开发过程中的 药物化学数据和知识;STITCH是一个用于检测化合物和蛋白质之间 相互作用关系的数据库。
(2)亲和力值怎么得到:
①Davis数据集是用Kd值取log作为亲和力值。
②对于KIBA数据集,它从多个来源(即Ki、Kd和IC50)集成到一个生物活性矩阵中,我们使用矩阵中的值(即KIBA值)作为亲和力值。
有关Kd,Ki,和IC50:
①Kd:(dissociation constant,解离常数),反映了蛋白质与小分子的亲和力,值越小亲和力越强。
②Ki:(inhibitor constant 抑制剂常数)针对的是蛋白质与抑制剂,反映的是抑制剂对靶标的抑制强度,值越小说明生物活性越高,抑制能力越强。
③IC50:半抑制浓度,达到50%抑制效果时抑制剂的浓度。半数抑制是用来衡量抗体灵敏度的。值越低,说明抗体的灵敏度越高。
(2)评估指标
①MSE:Mean Square Error,均方误差
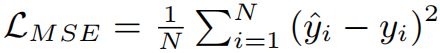
其中y^是预测值,y是真实值,N是药物对儿的数量。
②CI:Concordance Index,一致性指数
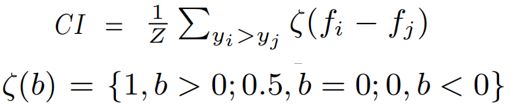
CI 一致性指数 用来检验 亲和力值 对 相应 药物-靶标相互作用的排序 是否与 基本事实 相同,计算方法是把数据集中的 所有研究对象 随机地两两组成对子。
Z是(不同亲和力值的)药物-靶标对数量。y是实际值,f是预测值。ζ(b)是一个阶跃函数(读作Zeta),ζ(b)=0或0.5时都代表模型没有起作用。
【e.g 两个药物-靶标对,如果效果较好的一对 其预测的 亲和力值 比 另外一对较高,那么预测结果与实际结果相符,ζ=1】
③r2m:
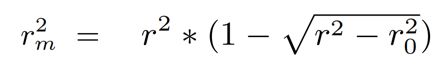
r2m用来评估(线性回归模型中)模型拟合程度,r和r0 分别是有无截距时的 真实值和预测值之间 的相关系数。越接近1效果越好。r2m可用于评估QSAR模型的 外部预测性能。
④AUPR:Area Under Precision Recall,PR曲线下的面积。
AUPR:PR曲线下的面积,PR曲线是召回率和正确率组成的曲线图。
AUPR作用:用来衡量不平衡数据集中模型的性能,值越大越好.
PR曲线横坐标是召回率(查全率)recall = TP/(TP + FN),即 对于所有的正例样本,有多少被预测出来了;
纵坐标是准确率(查准率)precision = TP/(TP+FP),即 所有预测为正例的样本,哪些预测对了。
(TP真正例,FP假正例,FN假反例)
(3)基准方法
①KronRLS:Kronecker Regularized Least Square
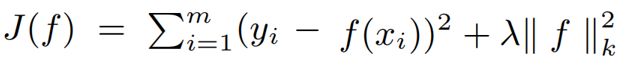
KronRLS方法:这方法基于Kronecker正则化最小二乘法。(用来最小化目标函数)。
xi 是输入(训练输入特征),yi 是真实值,f是非线性函数,λ是一个预定义的正则化参数,||f||2k 是以k为内核的范数。
②SimBoost
SimBoost模型:也是一个用来预测药物-靶标结合亲和力的模型,它使用了gradient boosting machine模型来表示输入特征与亲和力之间的非线性关系。
gradient boosting machine梯度提升机
简单理解:把预测的函数或模型看成参数
类比:神经网络中的梯度下降
方法:梯度下降
目的:每次迭代让损失减小。
③DeepCPI
将损失函数替换为MSE均方误差,输出层维度设为1。
DeepCPI模型最初专门用于DTI预测,它使用(基于r-radius指纹表示的)GNN来编码药物的分子结构,使用CNN来编码蛋白质序列,使用注意力机制来连接药物和蛋白质 进行预测。 在这里不能直接用于DTA任务,所以需要做一些改动。
④DeepDTA
DeepDTA以药物或者蛋白质序列的one-hot编码作为输入,分别针对药物和靶标训练两个CNN,分别从从SMILES串和蛋白质序列中学习特征,将学习到的特征连接起来并传到名为DeepDTA全连接层来预测亲和力。3个全连接层,层层之间引入dropout=0.1防拟合。
四、实验结果
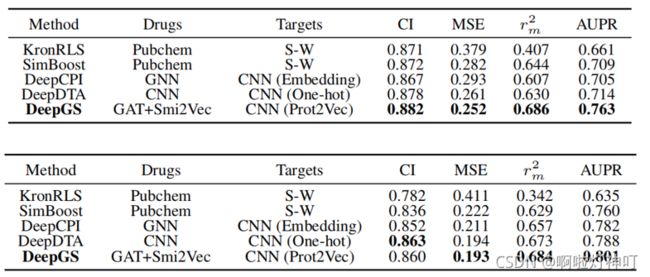
(1) 与其他模型比较
上图为在Davis数据集上是表现,下图是在KIBA数据集上的表现。
(1) 在Davis数据集上,DeepGS表现最好。
①基于深度学习的模型中,文章提出的DeepGS效果最好,原因有两个:Ⅰ与DeepCPI相比,DeepGS考虑了药物的拓扑结构和化学背景;Ⅱ 与DeepDTA相比,DeepGS既考虑了分子的拓扑结构,又使用了Smi2Vec和Prot2Vec,比DeepDTA的one-hot效果好。
这是因为one-hot比较稀疏,而且不能表示语意之间的关系。Smi2Vec和Prot2Vec通过embedding将 高维稀疏矩阵 转化为
低维稠密矩阵,而且具有相似语意的 映射在 空间的相似位置。
②KronRLS不如基于深度学习的模型效果要好,这是因为KronRLS等传统模型严重依赖于 手工设计的特征提取 以及 药物和靶标的 相似矩阵。
(2)在KIBA数据集上,DeepGS总体效果也是最好的,一致性指数CI略低于DeepDTA的原因是KIBA数据集的数据异质性(数据来源比较多样:Kd, Ki, IC50)。
(2) 预测值与实际值
模型在两个数据集上亲和力的预测值和实际值如图。越靠近 p = m 这条线,预测值和实际值越接近,说明模型效果越好。我们可以看出在两个数据集上,预测值和实际值都是比较接近的,说明模型有好的预测效果。
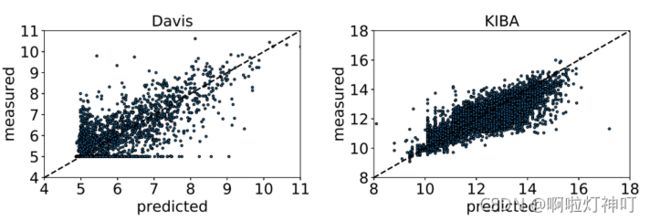
第一个图集中在5-6,是因为亲和力值为5的占数据集的一半以上。第二个图集中在10-14原因相同。
【Q】亲和力值怎么得到?
【A】①Davis数据集是用Kd值取log作为亲和力值。
②对于KIBA数据集,它从多个来源(即Ki、Kd和IC50)集成到一个生物活性矩阵中,我们使用矩阵中的值(即KIBA值)作为亲和力值。
(3) 模型灵敏度
灵敏度分析,就是改变模型(公式)的某个参数,引起这个模型输出的变化的程度。
方法:当训练BiGRU和CNN时,我们将输入序列的长度 固定在某个值。如果输入序列的长度大于l,我们将裁剪输入序列;如果小于l,则在输入序列的末尾使用零填充。
长度设置:对于药物的输入序列(SMILES串),将其长度设置为[50, 100, 500],如上图;(平均长度64)
对于蛋白质的输入序列(即氨基酸),将其长度设置为[500, 1000 2000],如下图。(平均长度788)
结果:①药物长度50与100,蛋白质长度500与1000,模型性能差距较大;②药物长度100与500,蛋白质长度1000与2000,模型性能差距较小。
原因:大多数SMILES串的长度在50-100之间,平均长度64。因此当SMILES串长度为100和500时,几乎不需要裁剪的SMILES串,SMILES串缺少的信息就比较少,性能差距很小;但长度为50时,可能需要裁剪较长的SMILES序列,导致性能下降。蛋白质序列长度集中在500-1000之间,平均长度788,原因是一样的。
这表明:当序列长度l 大于数据集中 序列平均长度 时,性能降低可能很小。

五、结论
本文提出了一个用来DTA预测的模型DeepGS,模型性能较好,其亮点如下:
(1)模型同时考虑了 药物的局部化学背景 和拓扑结构;
(2)使用了嵌入技术(Smi2V ec和PROT2Vec)将氨基酸和SMILES串编码成分布式表示。