单细胞测序学习笔记(二)——细胞注释
1.细胞鉴别
在上一步对各类细胞进行featureblot后,可以根据文献或者在线数据库来对细胞进行鉴别。此处我使用的是CellMarker网站。


对照起来进行鉴别,但是有些细胞簇有两个marker,有些细胞簇的marker有重合,所以鉴别这一步具有比较大的主观性,需要结合多个数据库或者文献进行判断。
在完成鉴别后,创建一个新的字符向量保存细胞名称,顺序从0-12一一对应。
以下是我自己的判断结果:
new.cluster.ids <- c("Memory T cell","CD14+ Mono", "CD8 T",
+ "NK", "CD8 T", "B", "B", "undefined", "Cytotoxic T", "NK","DC",
+"Megakaryocyte progenitor cell","CD1C+_B dendritic cell")接下来,使用names()函数,将pbmc的levels和new.cluster.ids一一对应。
names<- 是通用替换函数。The default methods get and set the "names" attribute of a vector (including a list) or pairlist.用levels中的字符向量设置new.cluster.ids的names属性。
如果levels()比names()短,则会以NA填充names()。
names(new.cluster.ids) <- levels(pbmc)
> new.cluster.ids
0 1 2
"Memory T cell" "CD14+ Mono" "CD8 T"
3 4 5
"NK" "CD8 T" "B"
6 7 8
"B" "undefined" "Cytotoxic T"
9 10 11
"NK" "DC" "Megakaryocyte progenitor cell"
12
"CD1C+_B dendritic cell"
可以看到对应关系
然后给pbmc中每个identity重命名
再次查看UMAP聚类图
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
设置NoLegend(),因为已经将名称添加到图上。
然后保存这一步结果
saveRDS(pbmc, file = "pbmc_final.rds")关于save和saveRDS,参考R语言 save与saveRDS - JoAnna_L - 博客园:
Rda与Rdata,前者是后者格式的缩写,在R里加载同样使用load()。
save与saveRDS的区别在于,前者保存的数据文件格式为.Rda或Rdata,读入R使用load(),载入的数据名称为原数据名称,不可指定。
后者保存的数据文件格式为.rds,读入使用readRDS,读入的数据名称可指定:name<-readRDS('.rds')。因此当保存单个对象时,推荐使用后者。
save不仅可以保存数据,也可保存模型结果、函数等,可保存至.R文件中。
2. 使用SingleR进行注释
SingleR是针对scRNAseq的一种注释工具,给出具有已知标签的样本(single-cell or bulk)的参考数据集,它基于与参考集的相似性来标记来自测试数据集的新的细胞。
首先调用一下需要的包,这里用了pacman包调用
p_load(SingleR,reshape2,celldex)接下来把pbmc的聚类结果单独储存为meta对象
meta <- [email protected]
获取标准化矩阵
关于GetAssayData()的用法# Get the data directly from an Assay object
GetAssayData(pbmc_small[["RNA"]], slot = "data")[1:5,1:5]
这一步是从Seurat对象的data槽中访问数据
pbmc_for_SingleR <- GetAssayData(pbmc, slot="data")参考seurat对象结构 - 简书:使用GetAssayData函数可以从Seurat对象访问数据。
可以使用SetAssayData将数据添加到counts,data或scale.data插槽中。新数据必须具有与当前数据相同顺序的相同细胞。添加到counts'或data`中的数据必须具有与当前数据相同的features。
下一步,使用SingleR里celldex的HumanPrimaryCellAtlasData对pbmc_for_SingleR里的数据进行注释,加上label
问题:
由于SingleR的数据库是通过ExperimentHub服务器下载的,大陆网络无法连接,会报错如下:
Cannot connect to ExperimentHub server, using 'localHub=TRUE' instead
Error in .updateHubDB(hub_bfc, .class, url, proxy, localHub) :
Invalid Cache: sqlite file
Hub has not been added to cache
Run again with 'localHub=FALSE'
在此处使用SingleR的解决方法参考了jimmy老师推荐的第二种方法:singleR的7个数据库文件下载失败的解决方案 - 云+社区 - 腾讯云
下载《生信会客厅》上传的数据库文件
链接:https://pan.baidu.com/s/13EjK-66tZKRsSl9G80S7uw
提取码:8mfb下载完成后里面有SingleR里的7个数据库的数据集合,我们在这里要用的 HumanPrimaryCellAtlasData在ref_Human_all的R.data文件当中
读取文件
pbmc.hesc <- SingleR(test = pbmc_for_SingleR, ref = refdata,
+ labels = refdata$label.main) 这里labels选项有label.main和label.fine,main大类注释,也可使用fine小类注释,不过小类注释准确性不好确定。
然后查看新注释的标签与seurat分类的结果的交叠情况
> table(pbmc.hesc[["labels"]],meta$seurat_clusters)
0 1 2 3 4 5 6 7 8 9 10 11 12
B_cell 0 0 0 0 0 264 265 0 0 0 0 0 2
CMP 0 0 0 3 0 0 0 0 0 0 0 0 0
GMP 0 0 0 0 0 0 0 0 0 0 0 0 1
Monocyte 2 922 0 0 0 1 0 0 0 104 51 0 12
NK_cell 0 0 1 327 46 0 1 3 0 0 0 0 0
Platelets 0 0 0 0 0 0 0 0 0 0 0 28 0
Pre-B_cell_CD34- 2 4 0 0 0 0 0 0 0 1 0 0 11
T_cells 1278 1 740 3 273 5 2 120 122 0 0 0 0可以看出来跟我自己的主观判断还是有所出入的
然后将注释后的结果做热图并保存为pdf
> pdf("plotScoreHeatmap.pdf")
> print(plotScoreHeatmap(pbmc.hesc))
> dev.off()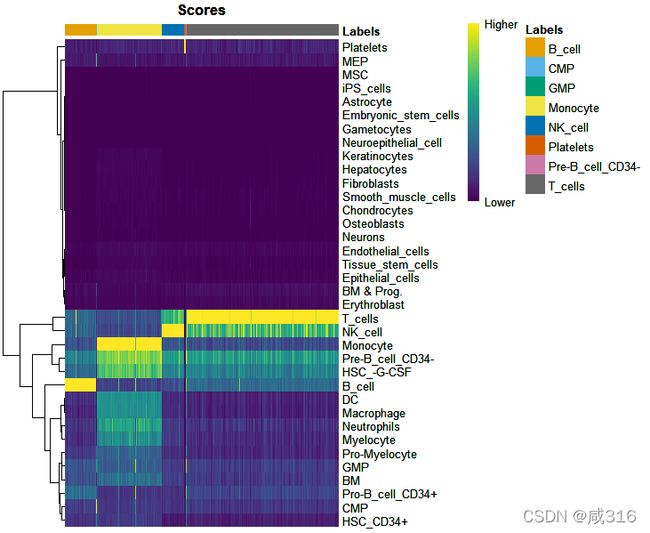
这样做出来的图比较漂亮和直观,能看到T细胞占了pbmc中大部分,其次是单核细胞
向pbmc_fianl的meda.data数据中加上pbmc.hesc的label(中间在运行的时候R关闭过一次,之后拿pbmc_final继续的分析,后续代码应该是pbmc的地方都换为pbmc_final)
[email protected]$labels <-pbmc.hesc$labels继续绘制DimPlot的对比图
print(DimPlot(pbmc_final, group.by = c("seurat_clusters", "labels"),
+ reduction = "umap", label = T))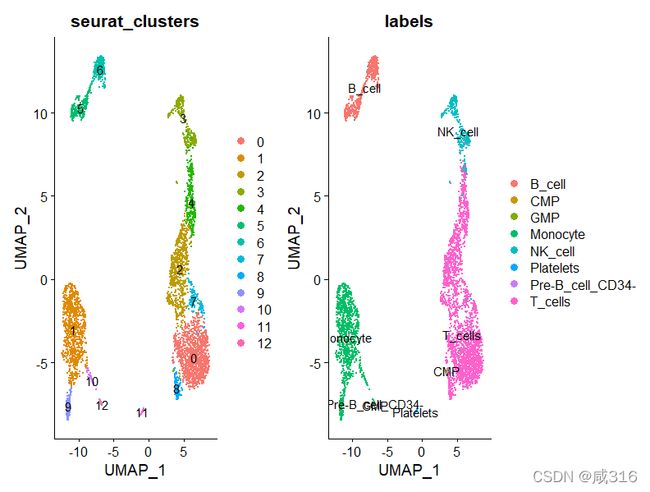
(这里右图label叠在一起了,除了用label.size缩小label字号以外还可以怎么修改)
更加美观的绘图方法如下:
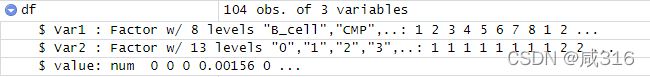
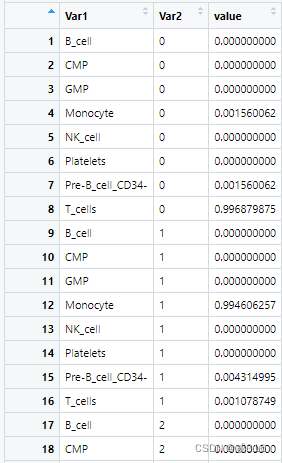
> aa=table(pbmc.hesc[["labels"]],meta$seurat_clusters)将labels信息和cluster信息合为列表存为aa
aa= apply(aa,2,function(x) x/sum(x))将aa值设置为百分比,apply()函数中2表示对aa中的数据按列(cell clusters)处理
df=as.data.frame(melt(aa))将aa转化为数据框
df$Var2=as.factor(df$Var2)把df中的Var2变量转化为因子(按照整数向量的形式储存类别值)
使用ggplot2绘制气泡图(上一步把Var2转化为因子)
> library(ggplot2)
> g <- ggplot(df, aes(Var2, Var1)) + geom_point(aes(size = value), colour = "green") + theme_bw()
> g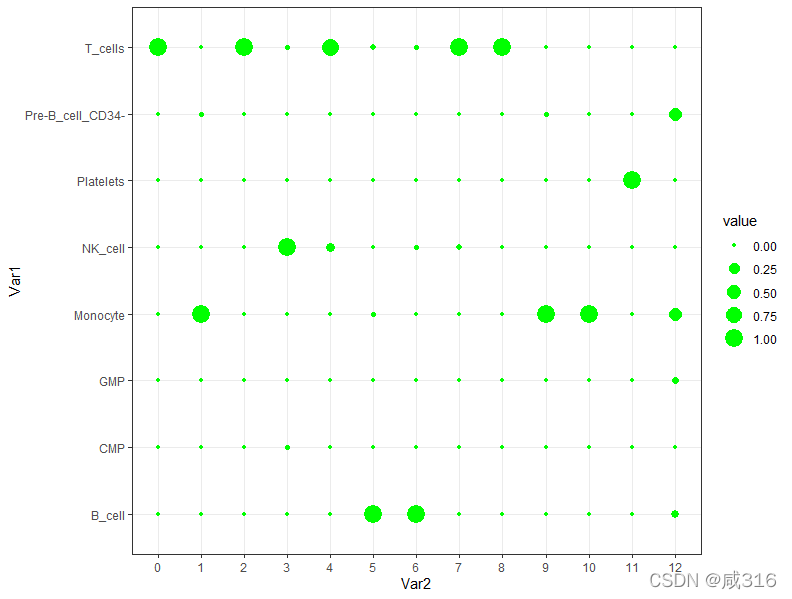
绘制热图
> library(pheatmap)
> pdf("heatmap.pdf",height=5,width=10)
> heatmap <- pheatmap(log2(aa+10), color=colorRampPalette(c("white", "blue"))(101))
> print(heatmap)
> dev.off()
