Geometry-enhanced molecular representation learning for property prediction|GeoGNN|将几何增强分子表示用于分子性质预测
这周读了一篇有关分子性质的文章《Geometry-enhanced molecular representation learning for property prediction》,文章于2022.2.7发表在 Nature Machine Intelligence 期刊上,期刊属于计算机 人工智能1区,3年平均IF为15.508。文章的创新点在于将3D信息(键长、键角、原子之间的距离)应用到分子表示当中。
文章源码:https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/pretrained_compound/ChemRL/GEM
采用的框架为 paddlepaddle。
1 Introduction
1.1 背景
随着DNN的发展,分子表征学习展现出了巨大的优势,其在分子性质预测方面的应用也越来越广泛,它将原子和键的拓扑结构视为一个图形,并将每个元素的信息传播到其邻域。
最近的研究开始在自监督方法中使用大规模未标记分子来预先训练分子表征,然后使用少量标记分子来微调模型。但现有的自监督技术只考虑分子的拓扑信息,忽略了分子的几何结构(3D结构)。
1.2 本文工作
本文提出了一种新的几何增强分子表征学习方法(GEM)。
①首先,为了使信息传递对几何体敏感,本文通过设计基于几何体的GNN体系结构(GeoGNN),同时对原子、键和键角的影响进行建模。
该体系结构由两个图组成:第一个图将原子视为节点,键视为边,而第二个图将键视为节点,键角视为边。
②其次,对GeoGNN进行预训练,从具有粗糙三维空间结构的大分子中学习化学定律和几何,设计各种几何级别的自监督学习任务。
③最后,为了验证所提出的GEM的有效性,在15个分子性质预测基准上将其与几个最先进的(SOTA)基线进行了比较,其中GEM获得了14个SOTA结果。
geometry-enhanced molecular representation learning method (GEM)
几何增强分子表示学习方法(GEM)
Geometry-based GNN architecture (GeoGNN)
基于几何的GNN结构(GeoGNN)
2 GEM框架
2.1 Geometry-based GNN architecture (GeoGNN)(对应3.2)
本文提出了GeoGNN,它通过模拟 原子-键-角度 关系来编码分子的几何结构信息。如图,左侧为atom–bond graph G=(V,ε),右侧为bond–angle graph H=(ε,A),其中V是原子集,ε是化学键集,A是键角集。
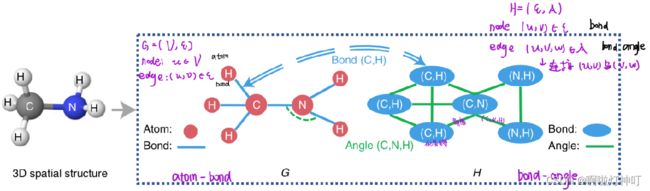
在原子键图G中,化学键被视为连接原子的边。
在键角图H中,键角被视为边,键角连接两个化学键和三个原子。
将 xu 作为原子u的初始特征,xuv 作为边(u,v)的初始特征,xuvw 作为键角(u,v,w)的初始特征。
将原子键图G和键角图H,原子特征、键特征和键角特征作为GeoGNN的输入,GeoGNN迭代学习原子和键的表示向量。对于第k次迭代,原子u和键(u,v)的表示向量分别用 hu 和 huv 表示。
为了连接原子键图G和键角图H,键的表示向量被视为G和H之间的通信链路。
step1:在键角图H中聚合相邻 键与键角 的信息来学习键的表示向量。
step2:在原子键图G中聚合相邻 原子与键 的消息来学习原子的表示向量。
step3:最后,通过聚合原子的表示来获得分子表示 hG。
(公式详见方法部分)
2.2 Geometry-level self-supervised learning tasks
为了进一步提高GeoGNN的泛化能力,本文提出了三个几何级的自监督学习任务来预训练GeoGNN,分别是:
(1)键长预测;
(2)键角预测;
(3)原子距离矩阵预测。
其中,键长和键角用于描述局部空间结构,而原子距离矩阵用于描述全局空间结构。
2.2.1 局部空间结构
键长和键角是最重要的分子几何参数。其中,键长是分子中两个连接的原子之间的距离,反映原子之间的键强度;键角是两个连续键(包括三个原子)的角度,描述分子的局部空间结构。为了学习局部空间结构,本文构建了预测键长和键角的自监督学习任务。
预测局部空间结构的任务可以看作是一个节点级的自监督学习任务。
step1:对于一个分子,随机选择15%的原子,对于每个选定的原子,提取该原子的一跳邻域,包括相邻的原子和键,以及该选定原子形成的键角;
step2:在一跳邻域中屏蔽这些原子、键和键角的特征。在GeoGNN的最终迭代中,提取的原子和键的表示向量 用于预测提取的键长与键角。
本文设计了一个回归损失函数来惩罚预测的键长/键角和标签之间的误差。(公式详见方法部分)
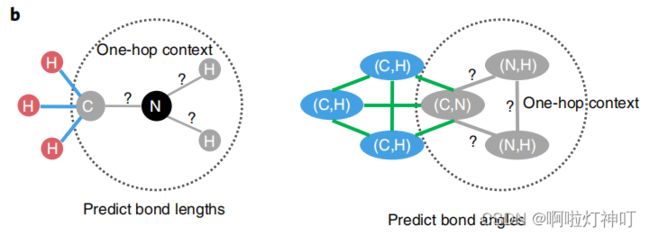
图:基于键长与键角的自监督学习任务
左边图G,右边图H。
图G中的黑色圆圈代表选定的原子,灰色圆圈代表相邻的屏蔽原子,灰色线条代表相邻的屏蔽键;
图H中的灰色椭圆也代表相邻的屏蔽键,灰色线条代表相邻的屏蔽键角。
2.2.2 全局空间结构
除了学习局部空间结构的任务外,本文还设计了学习全局分子几何的原子距离矩阵预测任务。
本文根据原子的三维坐标为每个分子构造原子距离矩阵,然后预测距离矩阵中的元素。注意,对于具有相同拓扑结构的两个分子,相应原子之间的空间距离可能会有很大差异。(损失函数在方法部分)

图:具有相同拓扑但不同几何形状的两种立体异构体之间的比较。对于左边的图, Cl-C-C 的角度为121°,对于右边的图, Cl-C-C 的角度为125°。
立体异构体:分子中原子或原子团互相连接次序及键合物质均相同,但空间排列不同而引起的异构体称为立体异构体。
因此,对于一个分子,本文没有将预测原子距离矩阵作为一个回归问题,而是将其作为一个多分类问题,通过以相等的步幅将原子距离投影到30个二进制表示中,也就是one-hot。有关设计损失函数的详细信息在方法部分。
为了预训练GeoGNN,本文通过总结相应的损失函数来考虑每个分子的局部空间结构和全局空间结构。
预测键长与原子距离的区别:预测键长的任务可以看作是预测原子距离任务的一个特例,但键长预测更关注局部空间结构,而原子距离更关注全局空间结构的分布。
3 方法
GNN是一种消息传递神经网络,给定节点v,它在第k次迭代中的表示向量 hv(k) 为:
消息传递神经网络,message passing neural networks,MPNN,作者将应用于图上的监督学习框架称之为消息传递神经网络

最后使用READOUT函数来整合节点信息,得到图的表示 hG :
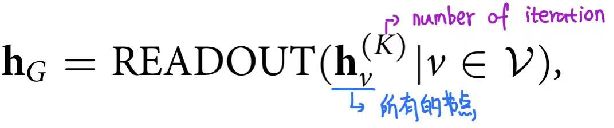
aggregate() 是用来聚合来自节点临域的消息的 聚合函数;
combine() 是用来更新节点表示的 更新函数。
readout是一个置换不变的池函数,比如求和 和 最大化
3.2 GeoGNN(对应2.1)
step1:在键角图H中聚合相邻 键与键角 的信息来学习键的表示向量。
键,也就是边(u,v),在第k次迭代中的表示向量 huv(k) 为:
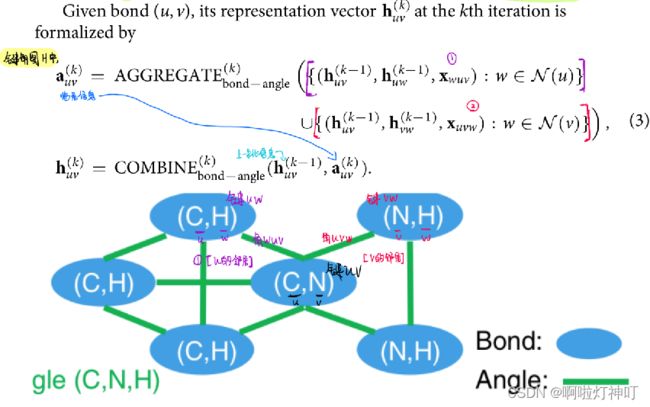
step2:在原子键图G中聚合相邻 原子与键 的消息来学习原子的表示向量。
原子u在第k次迭代中的表示向量 hu(k) 为:

键的信息 huv 是从键角图 H 中学习的,然后更新原子u的表示向量。
step3:在最后一次迭代的时候,通过聚集原子的表示来获得分子表示 hG,hG 用来预测分子的性质。

3.3 Geometry-level self-supervised learning tasks
3.3.1 局部空间结构
局部空间信息的自监督任务用于学习两个重要的分子几何参数:键长和键角。本文设计了一个回归损失函数来惩罚预测的 键长/键角 与 标签之间的误差,其定义如下:

3.3.2 全局空间结构
全局空间信息的自监督任务用于学习 所有原子对 之间的原子距离矩阵,距离矩阵中的每个元素都是两个原子之间的三维距离,用 duv 来表示分子中原子u和原子v之间的距离,损失函数如下:
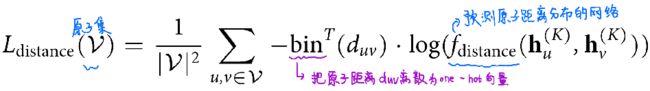
4 实验
4.1 Basic Setting
①数据集:
预训练:Zinc15
分子性质预测:MoleculeNet
②GNN结构:使用GIN中定义的聚合函数与组合函数,然后加上残差连接、层归一化、图归一化 来进一步提高性能,使用平均池化作为ReadOut来获取图表示。
③评估指标:
分类:ROC-AUC
回归:RMSE 与 MAE

RMSE(FreeSolv、ESOL和Lipo)
MAE:Mean Absolute Error平均绝对值误差,预测值与实际值的差值求绝对值,求和取平均(QM7、QM8和QM9)
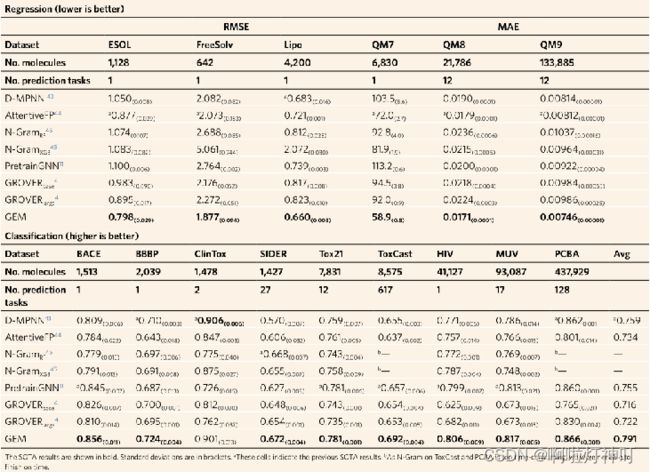
4.2 实验结果——①所有任务的总体性能
表格:
①最好的结果用黑体表示,括号内是偏差
②ToxCast 与 PCBA 上的N-Gram太耗时,无法按时完成
③回归的结果越小越好(因为是差值),分类的结果越大越好
结果解读:
①在15个数据集的14个中都取得了最先进的结果。
②在回归任务上,与之前最先进的结果相比,平均提升为8.8%;分类任务上的提升为4.7%。
③在分类任务上提升更大,可能是因为回归数据集侧重于预测 与分子几何形状高度相关的 量子化学性质。
模型:
D-MPNN:无pre-training;
AttentiveFP:无pre-training;
N-Gram:有pre-training。在图中以短距离行走的方式组装节点嵌入,然后利用随机森林或XGBoost来预测分子特性。
PretrainGNN:有pre-training。同时在节点水平和整个图的水平上预训练GNN,使GNN能够同时学习到有用的局部和全局表示。
GROVER base:有pre-training。通过两个自监督任务将GNN集成到Transformer中。base和large是不同的网络容量。
GEM:本文,geometry-enhanced molecular representation learning method ,几何增强分子表示学习方法
数据集:
回归:
ESOL::由实验水溶性数据组成。(由1128种水溶解度数据组成,直接从化合物的结构计算。化合物以SMILES表示,因此不包含分子中原子空间排列的信息)
FreeSolv:是水中水合自由能的数据集。(结构信息包含在SMILES字符串中)
Lipo:Lipophilicity,亲脂性数据集,指的是分子在非极性溶剂中的溶解能力。
分类:
BACE:BACE数据集提供了一组人类b-分泌酶1(BACE-1)抑制剂的定量(IC50)和定性(二元标记)结合结果
BBBP:血脑屏障渗透(Blood Brain Barrier Penetration,简称BBBP)数据集来自最近关于屏障渗透性建模和预测的研究。作为分隔循环血液和脑细胞外液的膜,血脑屏障可阻断大多数药物、激素和神经递质。因此,屏障的渗透在以中枢神经系统为靶点的药物开发中形成了一个长期存在的问题。该数据集包括超过2000种化合物的渗透性特性的二进制标签。
ClinTox:ClinTox数据集比较了FDA批准的药物和由于毒性原因未能通过临床试验的药物。该数据集包括1491种化学结构已知的药物化合物的两个分类任务:(1)临床试验毒性(或无毒性)和(2)FDA批准状态
SIDER:副作用资源(Side Effect Resource,简称SIDER)是已上市药物和药物不良反应(Adverse Drug Reactions,简称ADR)的数据库。DeepChem56中的SIDER数据集版本按照MedDRA(Medical Dictionary for Regulatory Activities,国际医学用语词典)分类,将药物副作用分为27个系统器官类别,对1427种已批准的药物进行了测量。
Tox21:“21世纪毒理学”(Toxicology 21,简称Tox21)倡议创建了一个测量化合物毒性的公共数据库,该数据库已用于2014年Tox21数据挑战赛。该数据集包含8014种化合物对12个不同目标的定性毒性测量,包括核受体和应激反应途径。
(12个endpoints,包括7个核受体信号和5个应激反应指标)
ToxCast:ToxCast来自与Tox21相同的计划,它为基于体外高通量筛选的大型化合物库提供毒理学数据。MoleculeNet中处理的集合包括8615种化合物的600多个实验的定性结果。
HIV:HIV数据集是由药物治疗计划(DTP)艾滋病抗病毒筛查引入的,该筛查测试了超过40000种化合物抑制HIV复制的能力。
MUV:Maximum Unbiased Validation (MUV) group是选自PubChem BioAssay的标杆数据集,使用提炼的最近邻分析。MUV数据集包括17个挑战任务,约9万个化合物,它用来验证虚拟筛选技术。
PCBA:PubChem BioAssay (PCBA)是一个包括高通量筛选得到的小分子生物活性的数据库。我们使用PCBA的子集,包括40万个化合物128个生物活性测定数据。
4.2 实验结果——②回归任务中不同GNN结构的性能
结果:①GeoGNN 在所有回归数据集上都大大优于其他 GNN 架构,与以往方法的最佳结果相比,整体相对提高了 7.9%;
②因为即使模拟了原子的三维坐标,GeoGNN 也包含几何参数。

多种GNN结构:(baseline)
(1)常用的GNN结构:GIN、GAT和GCN;
(2) 专为分子表示而设计的架构:D-MPNN、AttentiveFP 和 GTransformer;
(3) 使用三维分子几何结构的模型:SGCN、DimeNet 和 HMGNN 。
4.2 实验结果——③回归任务中不同预训练策略下GeoGNN的表现
为了研究所提出的几何级自监督学习任务的效果,采用不同类型的自监督学习任务对回归数据集上的GeoGNN进行预训练。
表格:“无预训练”表示无预训练的GeoGNN网络,“Geometry几何”表示我们提出的几何级别任务,“Graph图形”表示预测分子指纹的图形级别任务,“Context上下文”表示预测原子上下文的节点级别任务。
结果:①有几何级别任务的方法比没有几何级别任务的方法要好。
②“几何”在回归任务中的表现优于“几何+图形”,这可能是因为分子指纹与回归任务之间的联系比较弱。

5 结论
总结:现有的分子性质预测的预训练方法没有利用由键、键角和其他几何参数描述的分子几何结构,因此本文提出了基于几何的GNN结构(GeoGNN),并采用多几何层次的自监督学习方法来获取分子的空间知识。在多个数据集上进行了实验,GEM的表现优于其他基准方法。
创新点:提出并应用了基于键长与键角的自监督学习。