GEO数据挖掘(学习笔记)
GEO数据挖掘
- 1. GEO数据库简介
- 2. 数据下载
- 3. 数据质量检查
- 4. ID转换
- 5. 数据探索
-
- 5.1 PCA分析
- 5.2 hclust聚类
- 5.3 limma包进行差异分析
-
- 5.3.1 热图
- 5.3.2 火山图
- 5.4 GO and KEGG
生信菜鸟一枚,记录下学习生信技能树GEO数据挖掘相关视频的学习笔记。
1. GEO数据库简介
NCBI Gene Expression Omnibus(GEO)是一个存储各种高通量实验数据的公共数据库。这些数据包括基于单通道和双通道微阵列的实验,检测mRNA,基因组DNA和蛋白质丰度,以及非阵列技术,如基因表达系列分析(SAGE),质谱蛋白质组学数据和高通量测序数据。网址如下:https://www.ncbi.nlm.nih.gov/geo/
2. 数据下载
gset是一个列表,里面拥有表达矩阵、分组信息、临床资料等等内容,
###########################################
# GEO accession : GSE42872
# Platforms : GPL6244
# BioProject : PRJNA183688
##########################################
#####数据下载#####
if(!require(GEOquery)) BiocManager::install("GEOquery") # 安装包
package.version("GEOquery") # 查看版本
help(package = "GEOquery") # 查看GEOquery中的函数
library(GEOquery) # 加载包
library(tidyverse)
search() # 查看已加载R包
gset <- getGEO('GSE42872',destdir = ".",
AnnotGPL = F,getGPL = F)#下载GSE数据
save(gset,file = 'GSE42872.gset.Rdata')
# 读入表达矩阵,这里提供两种方法
exprset <- data.frame(exprs(gset[[1]])) # 推荐
exprset <- read.table(file = 'GSE42872_series_matrix.txt.gz',
sep = '\t',
header = T,
quote = '',
fill = T,
comment.char = "!",
check.names = T) #读取表达数据
rownames(exprset) = exprset[,1] #将第一列作为行名
exprset <- exprset[,-1] #去掉第一列
names(exprset) <- names(exprset) %>% substr(3,nchar(names(exprset))-1) # 更改列名
查看表达矩阵:


3. 数据质量检查
#####数据质量检查#####
head(exprset)
library(ggplot2)
library(dplyr)
library(reshape2)
ex = gset[[1]] # 实验相关信息
ph = pData(ex) # 每个样本的详细信息,包括了实验分组,芯片平台等
exprset_long <- melt(exprset) # 宽数据转长数据
# 从ph中可以看到实验分组信息
exprset_long$group <- rep(c("treat","control"),each = 3*nrow(exprset))
# {}代表创建了一个新环境,可以减少中间变量的产生
# 下面的代码依次产生了三幅图形
exprset_long %>% {
p <- ggplot(.,aes(variable,value,fill = group))
p1 <- p+ geom_boxplot()+
theme(axis.text.x = element_text(angle = 30,vjust = 0.6),
axis.title = element_blank())
p2 <- p+ geom_violin()+
theme_bw()+
theme(axis.text.x = element_text(angle = 30,vjust = 0.6),
axis.title = element_blank())
p3 <- ggplot(.,aes(value,col=variable)) +geom_density(lwd = 1)+
facet_wrap(~group,nrow = 2)+
theme_test()+
theme(axis.title = element_blank())
print(p1) # 这里如果只输入p1,则图形不会显示
print(p2)
print(p3)
# 依次产生箱线图,小提琴图和密度图
}
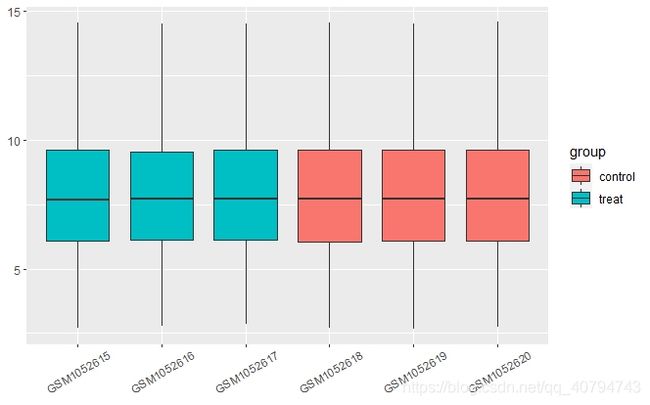
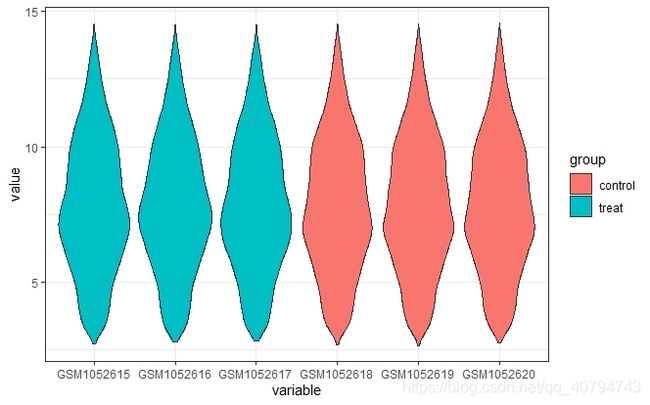
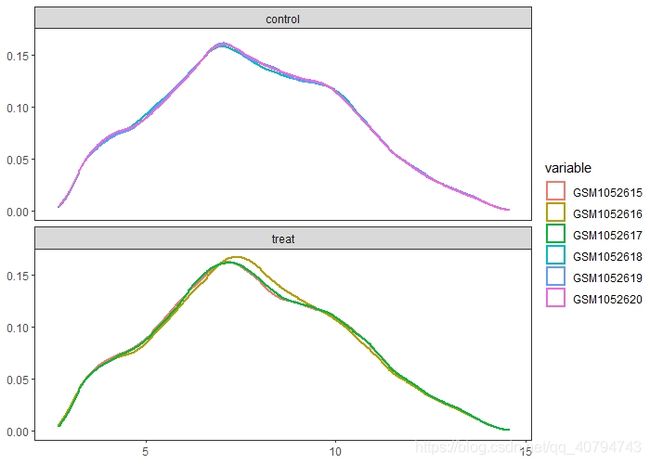
可以发现,这是已经标准化之后的数据,并且数据质量好,所以不必再进行标准化。
数据的质量检查完之后,刚开始我直接往下走了,结果发现表达矩阵中是探针ID,还好不是大的失误。在进行后续分析之前,需要进行ID转换。
4. ID转换
一般重要且常用的芯片平台在R的bioconductor里面都是有包的,可以参考生信菜鸟团的博文(http://www.bio-info-trainee.com/1399.html)。
#####ID转换#####
ex # 查看芯片平台,”GPL6244“
BiocManager::install("hugene10sttranscriptcluster.db")
library(hugene10sttranscriptcluster.db)
ls("package:hugene10sttranscriptcluster.db") # 查看包中的数据
symbol <- toTable(hugene10sttranscriptclusterSYMBOL) # 加载探针id和symbol的对应关系
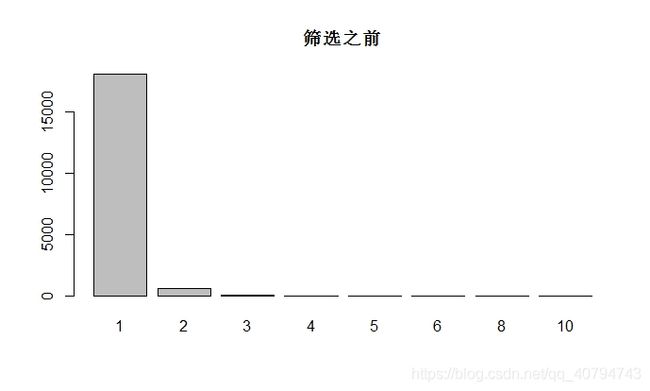
symbol$symbol %>% table() %>% table() %>% barplot(main = "筛选之前") # 查看探针和基因ID之间的对应关系
exprset.probe <- transform(exprset,probe_id = rownames(exprset))
exprset.symbol <- merge(exprset.probe,symbol,by = 'probe_id') # merge函数合并,初次筛选
exprset.symbol <- exprset.symbol[,c(8,2:7)]
exprset.symbol <- mutate(exprset.symbol,mean = rowMeans(exprset.symbol[2:7]))
exprset.symbol <- exprset.symbol %>% arrange(mean) %>%
distinct(symbol,.keep_all = T) %>% select(symbol:GSM1052620) # 依据symbol分组,依据mean排序,,之后再删除重复symbol
dim(exprset.symbol) # 查看数据维度
exprset.symbol$symbol %>% table() %>% table() %>% plot(main = "筛选之后") # 查看是否还有重复的symbol
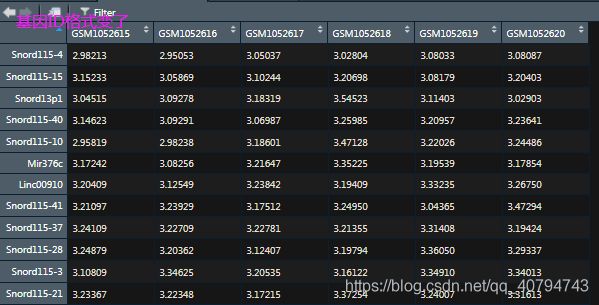
筛选后的表达矩阵:

5. 数据探索
对上一步的表达矩阵清洗
# 将gene symbol转换为首字母大写,其余小写的格式
id <- exprset.symbol$symbol %>% {paste(substr(.,1,1),tolower(substr(.,2,nchar(.))),sep = "")}
exprset.symbol <- mutate(exprset.symbol,symbol = id)
# 这里还有一种更为简便的方法,将gene symbol转换为首字母大写,其余小写的格式
# library(stringr)
# str_to_title(exprset.symbol$symbol)
# 将symbol设置为行名
rownames(exprset.symbol) <- exprset.symbol$symbol
exprset.symbol <- exprset.symbol[,-1]
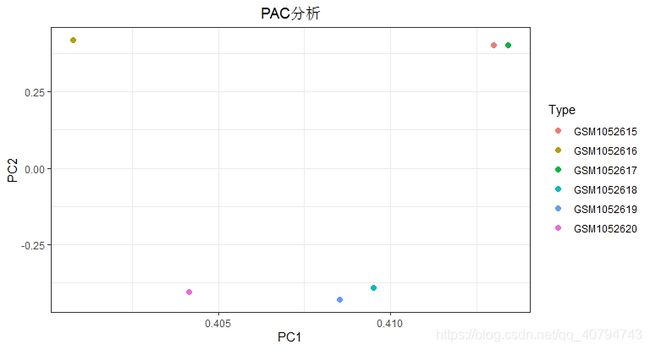
5.1 PCA分析
#####PCA#####
library(ggpubr)
pca.info <- prcomp(exprset.symbol)
summary(pca.info)
pca.data <- data.frame(sample = rownames(pca.info$rotation),
Type = colnames(exprset.symbol),
pca.info$rotation)
## 绘制PCA散点图
ggscatter(pca.data,x = "PC1",y = "PC2",
color = "Type",
size = 2
)+
theme_bw()+
ggtitle("PAC分析")+
theme(plot.title = element_text(hjust = 0.5))
5.2 hclust聚类
#####hclust聚类#####
exprset.symbol %>% t() %>%
dist() %>% hclust() %>%
plclust()

5.3 limma包进行差异分析
#####差异分析#####
library(limma)
group <- c(rep(c("control","treat"),each = 3))
design <- model.matrix(~0+factor(group))
colnames(design) = levels(factor(group))
rownames(design) = colnames(exprset.symbol)
contrast.matrix <- makeContrasts("treat-control", levels = design)
fit <- lmFit(exprset.symbol,design)
fit2 <- contrasts.fit(fit,contrast.matrix)
fit2 <- eBayes(fit2)
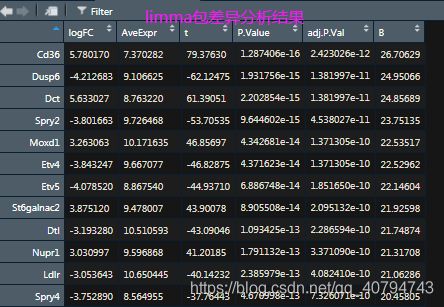
DEG_ot <- topTable(fit2, coef =1, n = Inf)
gpl42872.DEG = na.omit(DEG_ot)
head(gpl42872.DEG)
save(gpl42872.DEG,file = 'gpl42872.DEG.Rdata')
5.3.1 热图
#####热图#####
library(pheatmap)
choose_gene = head(rownames(gpl42872.DEG),25)
choose_matrix = exprset.symbol[choose_gene,]
choose_matrix = scale(choose_matrix)
pheatmap(choose_matrix,show_rownames = T)

5.3.2 火山图
#####火山图#####
logFC_cutoff = with(gpl42872.DEG,mean(abs(logFC))+2*sd(abs(logFC)))
gpl42872.DEG$change = as.factor(ifelse(gpl42872.DEG$P.Value < 0.05 &
abs(gpl42872.DEG$logFC) > logFC_cutoff,
ifelse(gpl42872.DEG$logFC > logFC_cutoff,
'up','down'),'no-change'))
gpl42872.DEG$logP <- -log10(gpl42872.DEG$P.Value)
library(ggplot2)
ggplot(data = gpl42872.DEG,
aes(x = logFC, y = -log10(P.Value),
color = change)) +
geom_point(size = 2) +
theme_bw() +
xlab("log2 fold change") +
ylab('-log10 p-value') +
scale_color_manual(values = c('blue','gray','red'))

5.4 GO and KEGG
GO和KEGG分析的平台很多,比如R语言的topGO包,DAVID网站等等。
#####GO and KEGG####
library(org.Hs.eg.db)
library(topGO)
library(clusterProfiler)
gene <- gpl42872.DEG %>% subset(P.Value < 0.05 & abs(logFC) > logFC_cutoff) %>% rownames()
gene.dt <- bitr(gene, fromType = 'SYMBOL',
toType = c('ENSEMBL','ENTREZID'),
OrgDb = org.Hs.eg.db)
dt <- gene.dt$ENTREZID
# GO
ego <- enrichGO(dt, #基因表
'org.Hs.eg.db' , #物种包
keyType = "ENTREZID",
ont="BP", #GO富集分析的分类,MF-molecular function ,BP-biological process ,CC-cellular compotent
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05,
pool = T,
readable = T
)
barplot(ego)
res<- as.data.frame( ego@result)
# KEGG
kegg<- enrichKEGG(dt,
organism = "hsa", #KEGG中物种缩写
pvalueCutoff = 0.05,
keyType = "kegg", #KEGG中编号格式
pAdjustMethod = "BH", #多重实验校正
qvalueCutoff = 0.2
)
barplot(kegg)
res<- as.data.frame( kegg@result)
GO和KEGG分析网上的教程不要太多,可以直接去生信菜鸟团找。