WGCNA 简明指南|2. 模块与性状关联分析并识别重要基因
WGCNA 简明指南|2. 模块与性状关联分析并识别重要基因
WGCNA 系列
WGCNA 简明指南|1. 基因共表达网络构建及模块识别
WGCNA 系列
参考
关联模块与临床特征
量化module-trait(模块-特征)关系
基因与性状和重要模块的关系:基因重要性和模块成员
模块内分析:鉴定高GS和MM基因
网络分析结果汇总输出
模块基因富集分析
往期
参考
本文主要参考官方指南Tutorials for WGCNA R package (ucla.edu),详细内容可参阅官方文档。
其它资料:
WGCNA - 文集 - 简书 (jianshu.com)
WGCNA分析,简单全面的最新教程 - 简书 (jianshu.com)
WGCNA:(加权共表达网络分析)_bioprogrammer-CSDN博客
WGCNA如何从module中挖掘关键基因_庐州月光的博客-CSDN博客
关联模块与临床特征
如果前面没保存再跑一遍WGCNA 简明指南|1. 基因共表达网络构建及模块识别以得到本次分析需要的数据。
量化module-trait(模块-特征)关系
在这个分析中,我们希望识别与临床特征显著相关的模块。由于我们已经有了每个模块的特征基因,我们简单地将特征基因与临床特征关联起来,并寻找最显著的关联。
Module eigengene
Module eigengene is defined as the first principal component of the expression matrix of the corresponding module.
# 导入之前的数据(也可以重新跑一遍上一期的内容)
lnames =load(file="FemaleLiver-02-networkConstruction-auto.RData");
lnames
# 明确基因和样本数
nGenes =ncol(datExpr);
nSamples =nrow(datExpr);
# 用颜色标签重新计算MEs
# 按照模块计算每个module的MEs(也就是该模块的第一主成分)
MEs0 = moduleEigengenes(datExpr, moduleColors)$eigengenes
# 对给定的(特征)向量重新排序,使相似的向量(通过相关性测量)彼此相邻。
MEs = orderMEs(MEs0)
# 计算基因模块MEs 与 临床特征的相关性以及p值
# use 给出在缺少值时计算协方差的方法
moduleTraitCor =cor(MEs, datTraits, use ="p");
moduleTraitPvalue = corPvalueStudent(moduleTraitCor, nSamples)可视化
# 如需保存
# pdf("Module-trait associations.pdf",width = 8, height=10)
par(mar =c(6, 8.5, 3, 3));
# 通过热图显示相关性
labeledHeatmap(Matrix = moduleTraitCor,
xLabels =names(datTraits),
yLabels =names(MEs),
ySymbols =names(MEs),
colorLabels = FALSE,
colors= greenWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text= 0.5,
zlim =c(-1,1),
main =paste("Module-traitrelationships"))
# dev.off()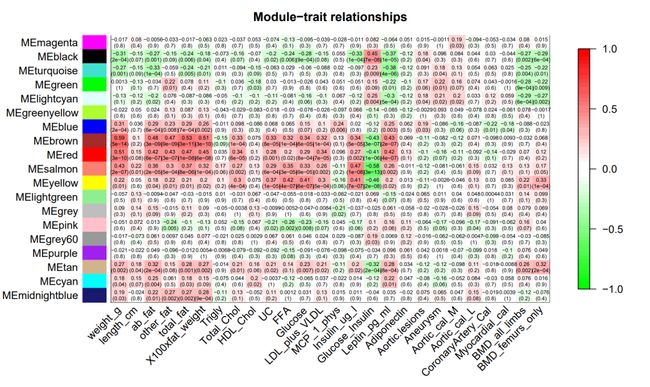 图1:模块-特征关联。每一行对应一个模块特征基因,每一列对应一个性状。每个单元格包含相应的相关性和p值。根据颜色图例,该表通过相关性进行颜色编码
图1:模块-特征关联。每一行对应一个模块特征基因,每一列对应一个性状。每个单元格包含相应的相关性和p值。根据颜色图例,该表通过相关性进行颜色编码
基因与性状和重要模块的关系:基因重要性和模块成员
Module Membership简称MM, 将该基因的表达量与module的第一主成分,即module eigengene进行相关性分析就可以得到MM值,所以MM值本质上是一个相关系数,如果基因和某个module的MM值为0,说明二者根本不相关,该基因不属于这个module; 如果MM的绝对值接近1,说明基因与该module相关性很高。
Gene Significance简称GS, 将该基因的表达量与对应的表型数值进行相关性分析,最终的相关系数的值就是GS, GS反映出基因表达量与表型数据的相关性。
# 以特征变量weight 为例
weight =as.data.frame(datTraits$weight_g);
names(weight) ="weight"
# 命名模块
modNames =substring(names(MEs), 3)
geneModuleMembership =as.data.frame(cor(datExpr, MEs, use ="p"));
MMPvalue =as.data.frame(corPvalueStudent(as.matrix(geneModuleMembership), nSamples));
names(geneModuleMembership) =paste("MM", modNames, sep="");
names(MMPvalue) =paste("p.MM", modNames, sep="");
geneTraitSignificance =as.data.frame(cor(datExpr, weight, use ="p"));
GSPvalue =as.data.frame(corPvalueStudent(as.matrix(geneTraitSignificance), nSamples));
names(geneTraitSignificance) =paste("GS.",names(weight), sep="");
names(GSPvalue) =paste("p.GS.",names(weight), sep="");模块内分析:鉴定高GS和MM基因
在感兴趣的基因模块中,筛选出高GS和MM的基因。在图1中可以看到,与性状weight_g相关性最高的基因模块是MEbrown,因此选择MEbrown进行后续分析,绘制brown模块中GS和MM的散点图。
module ="brown"
column =match(module, modNames);
moduleGenes = moduleColors==module;
sizeGrWindow(7, 7);
par(mfrow =c(1,1));
verboseScatterplot(abs(geneModuleMembership[moduleGenes, column]),
abs(geneTraitSignificance[moduleGenes, 1]),
xlab =paste("ModuleMembershipin", module,"module"),
ylab ="Genesignificanceforbodyweight",
main =paste("Modulemembershipvs.genesignificance\n"),
cex.main = 1.2, cex.lab = 1.2, cex.axis= 1.2,col= module)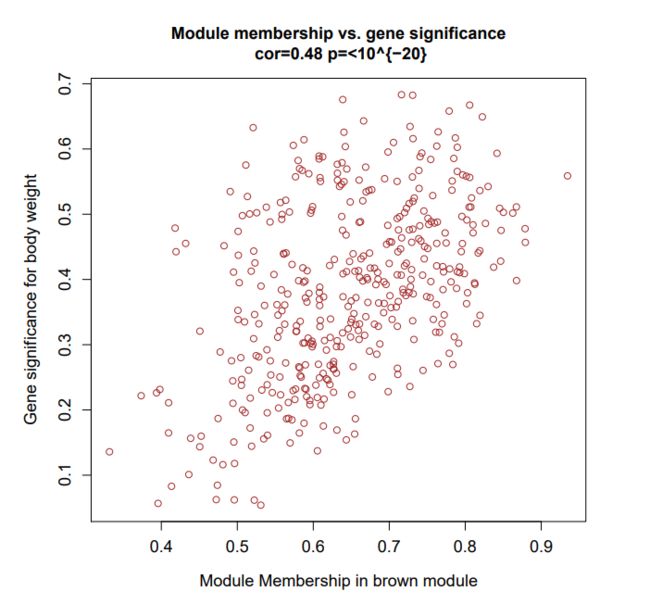 图2:棕色模块中weight与模块成员(MM)的基因显著性(GS)散点图。在这个模块中,GS和MM之间存在极显著的相关性
图2:棕色模块中weight与模块成员(MM)的基因显著性(GS)散点图。在这个模块中,GS和MM之间存在极显著的相关性
如图2所示,GS和MM是高度相关的,这说明与一个性状高度相关的基因往往也是与该性状相关的模块中最重要的(中心)元素。
网络分析结果汇总输出
我们已经找到了与我们的兴趣特征高度相关的模块,并通过MM确定了它们的核心参与者。现在,我们将这些统计信息与基因注释合并并导出。
names(datExpr)
# 返回brown模块中所有ID
names(datExpr)[moduleColors=="brown"]
# 导入注释文件
annot = read.csv(file = "GeneAnnotation.csv");
dim(annot)
names(annot)
probes = names(datExpr)
probes2annot = match(probes, annot$substanceBXH)
# 统计没有注释到的基因
sum(is.na(probes2annot))
# 创建数据集,包含探测ID ,
geneInfo0 = data.frame(substanceBXH = probes,
geneSymbol = annot$gene_symbol[probes2annot],
LocusLinkID = annot$LocusLinkID[probes2annot],
moduleColor = moduleColors,
geneTraitSignificance, GSPvalue)
# 通过显著性对模块进行排序
modOrder = order(-abs(cor(MEs, weight, use = "p")))
# 添加模块成员
for (mod in 1:ncol(geneModuleMembership))
{
oldNames = names(geneInfo0)
geneInfo0 = data.frame(geneInfo0, geneModuleMembership[,
modOrder[mod]],
MMPvalue[, modOrder[mod]]);
names(geneInfo0) = c(oldNames, paste("MM.",
modNames[modOrder[mod]], sep=""),
paste("p.MM.", modNames[modOrder[mod]], sep=""))
}
# 对基因进行排序
geneOrder = order(geneInfo0$moduleColor, abs(geneInfo0$GS.weight))
geneInfo = geneInfo0[geneOrder, ]
# 导出
write.csv(geneInfo, file = "geneInfo.csv")模块基因富集分析
批量导出模块基因后,使用在线网站或者R包clusterProfiler进行富集分析。
# Get the corresponding Locuis Link IDs
allLLIDs = annot$LocusLinkID[probes2annot];
# 选择感兴趣的模块
intModules = c("brown", "red", "salmon")
for (module in intModules)
{
# 模块探针
modGenes = (moduleColors==module)
# 得到 entrez ID
modLLIDs = allLLIDs[modGenes];
# 导出
fileName = paste("LocusLinkIDs-", module, ".txt", sep="");
write.table(as.data.frame(modLLIDs), file = fileName,
row.names = FALSE, col.names = FALSE)
}往期
跟着Nature学作图 | 配对哑铃图+分组拟合曲线+分类变量热图
(免费教程+代码领取)|跟着Cell学作图系列合集
跟着Nat Commun学作图 | 1.批量箱线图+散点+差异分析
跟着Nat Commun学作图 | 2.时间线图
跟着Nat Commun学作图 | 3.物种丰度堆积柱状图
跟着Nat Commun学作图 | 4.配对箱线图+差异分析
