单细胞测序系列之三:单细胞转录组测序
个人博客:https://www.horosama.com同步发布
相关文章链接:
单细胞测序系列之一:测序技术的发展
单细胞测序系列之二:单细胞基因组测序
文章目录
-
- 1. 单细胞转录组学的发展历程
- 2. 单细胞转录组学测序技术介绍
-
- 2.1 单细胞转录组学测序技术的开篇
- 2.2 单细胞转录组学测序的流程
-
- 2.2.1 单细胞悬液制备和单细胞分选
- 2.2.2 文库构建
-
- 2.2.2.1 Smart-seq2技术
- 2.2.2.2 10X Genomics技术
- 2.2.2.3 BD Rhapsody单细胞平台
- 2.2.3 生物信息学分析
- 3. 单细胞转录组学测序技术的对比和方法选择
- 参考文献
1. 单细胞转录组学的发展历程
单细胞转录组学测序技术是目前发展最为成熟的一个方向,和单细胞基因组学测序技术一样,其同样面临这样本量低而带来的一系列技术难题。一个人类细胞中,只有大约10 pg的总RNA,而其中mRNA的量只有0.2-0.3 pg左右,因此如何能够对如此微量的RNA进行建库同时去除大部分的mRNA就成了技术的核心关注点。
目前,单细胞转录组学测序技术的发展历程如下图所示。下面我们将对这些技术进行详细的介绍。

2. 单细胞转录组学测序技术介绍
2.1 单细胞转录组学测序技术的开篇
2006年,Kurimoto等发表了An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis这篇文章,其中使用的技术中,已经可见后来的单细胞测序技术的一些雏形1。
参见图2,在这篇文章中,其所使用的的技术步骤如下:
- 使用带有polyT尾的V1引物和带有polyA尾的mRNA结合进行逆转录,然后使用Exonucleas I去除多余的引物
- 通过末端转移酶(Terminal deoxynucleotidyl transferase,TdT)对上一步得到的cDNA末端加A,然后加入RNase H去除模板RNA链
- 以上一步得到的末端带A尾的cDNA作为模板,结合带有polyT尾巴的V3引物进行二链合成得到双链DNA
- 加入V1引物做20个cycle的PCR扩增
- 加入带T7 promoter的T7-V1引物做9个cycle引物
- 后续处理后使用microarray进行检测
2009年,汤富酬教授在Nature Methods发表了著名的单细胞转录组学测序开山之作:mRNA-Seq whole-transcriptome analysis of a single cell2,技术原理如图3所示:
- 首先在显微镜下人工吸取单个细胞并裂解
- 加入带有polyT尾的UP1引物进行逆转录得到cDNA分子
- 加入exonuclease I去除多余的引物,然后加入TdT对cDNA末端加A
- 加入带有polyT尾的UP2引物进行二联合成得到双链DNA
- PCR扩增后打断建库加上P1和P2接头,使用ABI的SOLID平台进行测序
对比2006年的Kurimoto的技术,我们可以看到其实二者十分的相似,最大的区别仅在于最后的检测手段不同,Kurimoto教授使用了microarray,而汤富酬教授用的是二代测序平台。
2.2 单细胞转录组学测序的流程
单细胞转录组测序一般可以分为单细胞悬液制备和单细胞分选,文库制备,高通量测序和生物信息学分析,数据可视化等几个步骤。
2.2.1 单细胞悬液制备和单细胞分选
对于大多数类型的样本来说,单细胞悬液的制备不算困难,因此不做过多的介绍。
单细胞分选有多种方法,参见图4包括pipetitte、microscope、LCM、microfluidics和FACS等,需要根据具体的使用场景来选择最合适的方法。不同的方法之间的对比可以参考下面的表格:
| 分选方法 | 操作原理 | 可分选细胞数量 | 分选效率 | 适用场景 |
|---|---|---|---|---|
| pipetitte | 使用移液管进行反复梯度稀释 | 少 | 低 | 适用于样本较少时 |
| microscope | 在显微镜下使用毛细管吸取细胞 | 少 | 低 | 分选早期胚胎等 |
| FACS | 流式荧光细胞分选 | 多 | 高 | 最常用的方法之一 |
| LCM | 使用激光系统从固体样本中分离细胞 | 少 | 低 | 适用于福尔马林固定、 冷冻固定或石蜡包埋的 固体样本 |
| microfluidics | 微流控系统分选 | 多 | 高 | 样本消耗低、但对介质 固体污染比较敏感 |
| CTC富集 | 使用抗体结合的磁珠抓取CTC细胞 | 少 | 高 | 分离CTC细胞 |
2.2.2 文库构建
单细胞RNA测序建库技术种类繁多,截至目前已经有了一百多个了。参考图5我们可以发现,从2009年开始,单细胞转录组测序技术的发展速度非常快,每年都有新的技术出现。但一直到2014年,单细胞技术的通量还停留在100个细胞以下,即使是当时最为先进的Fluidigm C1平台也是如此。
Fluidigm C1平台,是最早被应用于single cell领域的商业化分选技术,基于Fluidigm的微流控技术,大约在几个小时中可以捕获96个左右的细胞,进行各类的single cell测序建库。
在2014-2015年间,几个重磅级的技术CytoSeq、Drop-Seq、inDrop技术出现了,将单细胞技术的通量提高到了10000-100000个细胞的量级。而在2017-2018年,10X Genomics和BD Rhapsody的问世,使得单细胞技术真正的得以进入“大众化”阶段。

如此多的测序技术,想要全部介绍完自然是不现实的,所以接下来,我们将挑选几个最常用和最常见的单细胞转录组测序技术平台进行介绍,分别是Smart-seq2,10X Genomics和BD Rhapsody。对于目前的各种技术的对比可以参考图6,其中我们最需要关注的点是技术的转录本覆盖度和细胞通量,比如当我们需要检测splicing variants的时候,就需要选择full length的Smart-seq2,更为关注转录起始点(TSS)时就需要选择5’ end的测序方法,而需要做大规模的单细胞检测时,Drop-Seq和SPLIT-seq等技术就更为合适。

2.2.2.1 Smart-seq2技术
Smart-seq于2012年由Rickard Sandberg教授发明3。从前面对汤富酬教授的技术介绍我们可看出,基于末端加A的方法有明显的3’末端偏好,而SMART-seq通过使用模板转换的原理(template switch),可以更好的扩增全长RNA,对转录本的测序覆盖度明显提高。
2013年,Rickard Sandberg教授在Smart-seq的基础上改良发明了Smart-seq2技术4,其具有高灵敏度、低偏好性的cDNA扩增,更高的转录本的整体覆盖率。多年以来,Smart-seq2技术一直被视为单细胞转录组测序领域的“金标准”。
Smart-seq2的原理图如图7所示,其主要步骤如下:
- 使用oligo(dT) primer对带有polyA尾的RNA进行反转录,其中使用的逆转录酶为MMLV(Moloney Murine Leukemia Virus)逆转录酶,MMLV逆转录酶具有末端转移酶活性,会在cDNA链3’端加上三个C
- 加入模板转换引物TSO(template-switching oligo),TSO 3’端有三个G能与一链3’端的三个C互补,而且最末端的+G是做了锁核酸修饰的G,能增加TSO的热稳定性,以及其与一链cDNA游离的3’端的互补的能力,TSO还带有PCR所需的引物(图中绿色的部分)。TSO引导MMLV逆转录酶发生模板转换,使其继续逆转录TSO序列获得双链cDNA。
- 加入PCR引物进行PCR预扩增
- 利用Tn5转座酶对DNA进行打断的同时将接头添加到cDNA的两端,再进行最后一次PCR扩增后,即可上机测序。
相比较于Smart-seq,Smart-seq2的主要技术优化点包括5:
- 使用了更温和的细胞裂解液来避免对后续酶反应的影响
- 加入甜菜碱和Mg2+提高逆转录酶的热稳定性和反转效率
- 甜菜碱(N,N,N三甲基甘氨酸)是一种高效的甲基供体。在反转录体系中加入甜菜碱,其能够增加转录所需酶(蛋白质)的热稳定性;一些RNA有着例如发夹结构、环形结构等二级结构,影响逆转录酶与其结合,加入甜菜碱可解决这一问题,最终得到完整的cDNA文库。此外,在加入甜菜碱的同时增加Mg2+的浓度也可在一定程度上提高cDNA产量。
- 加入10个额外的加热循环(50°C 2min;42°C 2min)来解开RNA二级结构。
- oligo-dT引物与TSO引物的末端序列一样,这样在扩增双链cDNA只需要一种引物
- TSO末端的最后一个G使用了锁核酸修饰
- 锁核酸修饰是通过在核糖上加入一个2’-O,4’-C-亚甲基桥链,把糖环的空间构象锁定,提高核酸互补链结合的稳定性,进而提高链结合的特异性。
Smart-Seq2对转录本覆盖度很高,可以做到50 pg RNA起始建库,可mapping reads的比例高。但其也有着自己的缺点,比如建库过程非链特异,对长转录本的建库效率较低,对于高丰度转录本存在PCR偏好和细胞分选效率较低等。
2.2.2.2 10X Genomics技术
10X Genomics的技术原理和Drop-Seq相似,是目前单细胞转录组测序最流行的技术平台之一,可以实现大规模的单细胞转录组测序,具有细胞通量高、细胞捕获率高和项目周期短等优点。10X Genomics一次测序可以捕捉100-80,000个细胞,具有极高的细胞通量。
参考图8,10X Genomics的工作流程包括:单细胞悬液制备,细胞标记和文库制备,测序,数据分析和结果可视化。

10X Genomics的技术关键点之一在于其凝胶微珠Gel Beads的设计,每个凝胶微珠上有40-80万个特定的核酸引物序列,序列包括如下几个部分:
- TruSeq Read 1:测序引物
- 10X Barcode序列:共16 nt,每个Gel Beads对应一种Barcode,用来区分不同的细胞
- UMI(Unique Molecular Identifier):共12 nt,用来区分同一细胞中的每一个RNA分子
- poly(dT):用于捕获mRNA

相比于其它技术,主要的区分点在于第二步的Barcoding&Library Construction,其流程如下:
- 细胞分选:使用微流控技术对细胞进行分选,横向孔道逐个输送带有barcode的Gel beads,第一个纵向道输入细胞和酶,细胞会被吸附在微珠上,然后运送到第二个纵向通道和油相混合形成油包水结构的GEMs(Gel Bead in Emulsion)。
- GEMs形成后,细胞裂解,Gel Beads会自动溶解释放出Barcoode序列,这些序列和mRNA结合后逆转录得到cDNA。因为GEMs的存在使不同细胞的反应被物理隔离,保证了每个细胞对应了独一无二的Barcdoe。
- 将同一管内的cDNA混合后进行标准建库,得到的文库就可以进行测序。
- 得到数据后,使用Cell Ranger等软件进行数据分析,Cell Ranger是10X Genomics提供的一个软件包,可以直接输入Illumina测序的元素数据后输出表达定量矩阵,进行PCA,聚类和t-SNE/UMAP可视化等分析。
目前单细胞的降维图大部分选择使用UMAP(Uniform Manifold Approximation and Projection),因为其可以在最大程度保留原始数据的特征。一个典型的UMAP降维图如图10所示。

更加详细的介绍请参考下面的视频。
10x Genomics Single Cell Gene Expression测序
2.2.2.3 BD Rhapsody单细胞平台
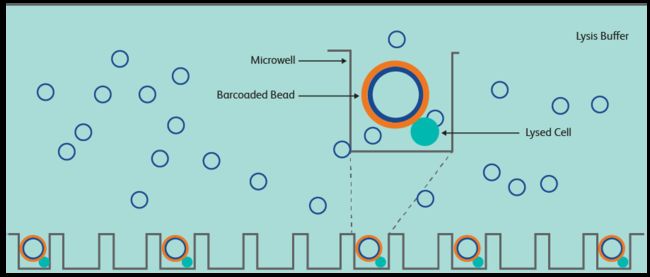
BD Rhapsody是基于微孔技术(microwell)的单细胞平台,其技术原理来自于2015年Science上发表的Cyto-Seq技术6,使用铺满20万个直径50 μm的微孔板和直径35 μm的beads来捕获细胞。
将单细胞悬液铺到微孔板上之后,细胞沉降到微孔底部,通过控制悬液的细胞密度来减少一个微孔捕获多个细胞的情况。然后将beads铺到微孔板上,加入细胞裂解液后,细胞释放出的mRNA就可以被beads上的序列在微孔内捕获,回收beads就可以进行后续的逆转录和建库等操作。Beads上的序列设计和上面提到的gel beads上的序列设计类似,这里不再赘述。
BD Rhapsody配套的扫描仪可以使得每一步操作时,微孔板内的状态可观察,能够随时进行质控和实验调整,而且其捕获效率最高可达97%。
2.2.3 生物信息学分析
单细胞转录组测序数据分析的工具和方法比较多,2020年Nature Protocols上发表的Tutorial: guidelines for the computational analysis of single-cell RNA sequencing data一文详细介绍了单细胞RNA-seq数据的一般分析流程,可以作为重要的参考7。这里将对这边文章进行简单的介绍,有兴趣的可以下载文章进行详细的阅读。
单细胞RNA-seq数据分析的核心是表达矩阵,表达矩阵代表了每个细胞中每个基因的转录本的检测到的数量。整个单细胞RNA-seq数据分析的流程可以分成两个主要的部分:生成表达矩阵和分析表达矩阵。因为通常单细胞测序的样本起始量较低,测序深度也不高,很多基因可能不能被检测到,这导致了在表达矩阵中存在着非常高比例的零值,即使是高深度测序的数据集中也可能有约50%比例的零值,一些低深度测序的数据中甚至99%都是零值。如何区分和处理这些零值是单细胞数据分析中的主要挑战。
文章将单细胞RNA测序数据分析的流程分为了以下几个步骤:
-
Quality control
- 去除不太可能代表完整的单个细胞的barcode,比如对于每一个barcode设置UMI的最低阈值
- 区分完整的活细胞和受损或者垂死的细胞,具有较高比例的线粒体相关基因、检测到的基因数过少或者不能比对/多重比对的reads占比过高的往往代表了垂死或者受损细胞
- 区分一个barcode对应了多个细胞的情况,可以通过scrublet或者DoubletFinder等软件来分析
-
Normalization
- SCnorm通常用于低通量、高深度的数据集
- sctransform用于高通量、低深度的数据集
- 基于贝叶斯分析的bayNorm可以在考虑到mRNA捕获效率的情况下进行表达矩阵推断
-
Batch effect correction
- 对于bulk seq,可以使用ComBat来去除批次效应
- 可以使用mnnCorrect来去除单细胞批次效应,该方法基于MNN(mutual nearest neighbors)算法。这种方法也被使用典型相关性分析CCA(canonical correlation analysis)算法的Seurat来寻找锚点。
- 可以使用深度学习算法来进行矫正,比如MMD-ResNet,scGen和华大开发的deepMNN等。
-
Imputation and smoothing
- 有多种方法可以被用来填补零值,比如scImputer,DrImpute和SAVER等,这些工具依赖于寻找数据中心可以被用来预测缺失值的结构特征。
- MAGIC和scVI方法应用了平滑算法来减少噪声。
- SAVER-X和netNMF-sc通过使用外部参照来填补缺失值。
-
Cell cycle assignment
- cyclone和Seurat是两个广泛使用的用来识别细胞周期的工具。
-
Feature selection
- 单细胞数据中每个基因代表一个维度,因此一个人类数据集可能有高达20000个维度
- 特征选择用于将具有最强的生物学信号的特征从背景中分离出来。最常使用的方法是考虑寻找反差高于预期的基因。
- Seurat使用了非参检验的方法来识别特征,而对于罕见细胞类型中的差异表达的基因,可以使用GiniClust方法。
-
Dimensionality reduction and visualization
- 过高的维度会带来“维度灾难”,减少高纬度带来的负面作用的一个方法是进行降维分析。
- 最常用的方法是主成分分析(principal component analysis,PCA)
- UMAP是目前最常用的可视化算法,并且取代了t-SNE,因为UMAP可以更好地保留数据的高维数据结构。
-
Unsupervised clustering
- k-means是最常用的聚类算法,是单细胞一致性聚类算法(single-cell consensus
clustering ,SC3)的基础。 - 另一个例子是基于层次聚类的Louvain算法,被Seurat和scanpy所采用的
- k-means是最常用的聚类算法,是单细胞一致性聚类算法(single-cell consensus
-
Pseudotime
- 基于单细胞表达谱,推断出哪些细胞类型向另外一种细胞类型发育的轨迹被称为拟时序分析。
- 一种方法是使用降维技术来识别细胞所处的低维流行,利用细胞和细胞间的连线图来表示轨迹,比如Monocle和DPT软件。
- 一种方法是利用无监督聚类先将不同的细胞类型进行聚类,然后连接每一个聚类,再把单独的细胞投影到分枝上,比如TSCAN和Mpath软件。
- 还可以使用RNA降解速率来推测细胞发育轨迹,比如RNAvelocity。
-
Differential expression
- 通常使用非参数Wilcoxon test来做检验
-
Comparing versus combining datasets
- 可以使用scmap或者MetaNeighbor来对数据集进行比较和合并。
文章里还列出了不同的任务中一些最常用的方法和方法所基于的理论依据。

3. 单细胞转录组学测序技术的对比和方法选择
最后,有如此之多的单细胞转录组学测序方法,我们如何来进行选择呢?一般来说,这需要根据我们所关心的生物学问题来决定。比如,基于droplet的方法更适合于对组织异质性进行分析,而关心不同的RNA isoforms的研究会去选择full length的Smart-seq2等方法。
2020年一项研究对比了不同的高通量和低通量的单细胞转录组学方法,从图12我们可以看出低通量的方法有这更高的灵敏度,可以检测到更多的UMI和基因数目8。
更多的方法之间的对比可以参见图14。
参考文献
Kurimoto, K., Yabuta, Y., Ohinata, Y., Ono, Y., Uno, K. D., Yamada, R. G., Ueda, H. R., & Saitou, M. (2006). An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis. Nucleic acidxis research, 34(5), e42. https://doi.org/10.1093/nar/gkl050 ↩︎
Tang, F., Barbacioru, C., Wang, Y., Nordman, E., Lee, C., Xu, N., Wang, X., Bodeau, J., Tuch, B. B., Siddiqui, A., Lao, K., & Surani, M. A. (2009). mRNA-Seq whole-transcriptome analysis of a single cell. Nature methods, 6(5), 377–382. https://doi.org/10.1038/nmeth.1315 ↩︎
Ramsköld, D., Luo, S., Wang, Y. C., Li, R., Deng, Q., Faridani, O. R., Daniels, G. A., Khrebtukova, I., Loring, J. F., Laurent, L. C., Schroth, G. P., & Sandberg, R. (2012). Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nature biotechnology, 30(8), 777–782. https://doi.org/10.1038/nbt.2282 ↩︎
Picelli, S., Björklund, Å. K., Faridani, O. R., Sagasser, S., Winberg, G., & Sandberg, R. (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nature methods, 10(11), 1096–1098. https://doi.org/10.1038/nmeth.2639 ↩︎
单细胞转录组方法篇——下 ↩︎
Fan, H. C., Fu, G. K., & Fodor, S. P. (2015). Expression profiling. Combinatorial labeling of single cells for gene expression cytometry. Science (New York, N.Y.), 347(6222), 1258367. https://doi.org/10.1126/science.1258367 ↩︎
Andrews, T. S., Kiselev, V. Y., McCarthy, D., & Hemberg, M. (2021). Tutorial: guidelines for the computational analysis of single-cell RNA sequencing data. Nature protocols, 16(1), 1–9. https://doi.org/10.1038/s41596-020-00409-w ↩︎
Ding, J., Adiconis, X., Simmons, S. K., Kowalczyk, M. S., Hession, C. C., Marjanovic, N. D., Hughes, T. K., Wadsworth, M. H., Burks, T., Nguyen, L. T., Kwon, J., Barak, B., Ge, W., Kedaigle, A. J., Carroll, S., Li, S., Hacohen, N., Rozenblatt-Rosen, O., Shalek, A. K., Villani, A. C., … Levin, J. Z. (2020). Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nature biotechnology, 38(6), 737–746. https://doi.org/10.1038/s41587-020-0465-8 ↩︎