2021-06-13
论文解读:《基于图形卷积网络,通过特征图和拓扑图进行图形采样,识别微小核糖核酸相关疾病》
- 一、摘要
- 二、简介
- 三、相关工作
- 四、材料和方法
- 五、节点特征构造
- 六、方法
- 七、结果和讨论
- 八、案例研究
- 九、结论
文章地址:https://academic.oup.com/bib/advance-article-abstract/doi/10.1093/bib/bbab165/6261915?redirectedFrom=fulltext
DOI:https://doi.org/10.1093/bib/bbab165
期刊:Briefings in Bioinformatics(2区)
发布时间:2021年4月2日
补充数据:https://academic.oup.com/bib.
代码:https://github.com/khanhlee/bert-enhancer
一、摘要
准确识别微小核糖核酸-疾病关联有助于理解各种疾病的病因和机制。然而,实验方法既昂贵又耗时。因此,迫切需要发展面向计量吸入器预测的计算方法。基于图论,本文将主成分分析预测作为一项节点分类任务。为了解决这个问题,我们提出了一种新的方法——图卷积神经网络模型预测方法,该方法通过特征图和拓扑图的图抽样来预测基于图卷积神经网络的模型,以提高训练效率和精度。该方法对特征空间的潜在联系和主成分分析数据的结构关系进行建模。图的节点由疾病语义相似度、微小核糖核酸功能相似度和高斯相互作用轮廓核相似度表示。此外,我们首次在MDA预测问题上同时考虑了6项任务,确保在均衡和非均衡样本分布下,MDA-GCNFTG不仅可以预测新的MDA,而且可以预测没有已知相关miRNAs的新疾病和没有已知相关疾病的新miRNA。五倍交叉验证的结果表明,MDA-GCNFTG方法在所有六个任务上都取得了令人满意的性能,明显优于经典的机器学习方法和最先进的MDA预测方法。此外,还通过图抽样策略和主成分分析-遗传神经网络拓扑结构图验证了遗传神经网络的有效性。更重要的是,对两种疾病和三种微小核糖核酸进行了案例研究,取得了令人满意的结果。
二、简介
MiRNA是1993年发现的一种内源性调控非编码RNA,长度约为22个核苷酸[1,2]。它通过靶向特定的mRNA和调节基因表达[3–7],在多种生物过程中发挥重要作用,包括免疫反应[8]、细胞周期调节[9]、肿瘤侵袭[10]等。此外,已证明miRNAs调节超过三分之一的基因[11],因此miRNAs的失调可导致细胞行为障碍[12]。此外,许多研究证明,微小核糖核酸与复杂的人类疾病的发展高度相关[13-16],特别是癌症[17],如乳腺癌[18,19],肺癌[20,21],淋巴瘤[22]等。因此,微小核糖核酸可用作疾病诊断中的潜在生物标志物[19,23,24]。因此,确定微小核糖核酸和疾病之间的联系不仅可以提高对疾病机制的理解,而且有助于疾病的预防、诊断和治疗[25,26]。尽管鉴定微小核糖核酸-疾病关联的实验方法具有很高的准确性,但它们非常耗时且昂贵。因此,发展计算方法来确定计量吸入器是必要的,并成为实验方法的一个辅助步骤[27]。
网络科学是探索复杂生物系统(即分子相互作用网络)的主干。它们是由作为节点的生物分子和作为边的生物分子之间的互连组成的图,例如在本工作中研究的MDA。大量研究表明,生物分子并不单独发挥其生物学功能,而是通过与其他生物分子的相互作用来表达其功能,从而形成一个分级的群落结构[28]。此外,该疾病应被描述为“网络疾病”,因为它很少是由单一基因异常引起的,而是由组织和器官系统的复杂生物网络的干扰或故障引起的[29]。因此,生物分子间关联的推断要考虑网络拓扑。图形神经网络(GNNs) [30]代表了直接在网络/图形结构数据上操作的重大进步,以及解决上述问题的有前途的方法。GNN本质上是一种邻域节点聚合方案,其中每个节点聚合其有向邻居的特征信息来计算其新的特征向量。在信息聚合的多次迭代之后,计算的节点嵌入将捕获节点的邻居之间的结构信息。GNN被广泛应用于各种实际任务中,并在生物信息学应用中取得了令人满意的性能,例如药物-靶相互作用或亲和力预测[31–36],药物-药物相互作用预测[37–40],疾病-基因关联鉴定[41–44]等。
图卷积网络(GCN) [45]是GNN的一个重要分支,近年来取得了很大进展。然而,传统的GCN方法通常需要全图训练。在MDA或其他生物信息学任务中,相关实体(如药物、蛋白质、miRNAs等)的数量。)很大。因此,盲目地执行全图训练将由于“邻居爆炸”现象而导致巨大的计算复杂性,并且可能由于需要太多的计算资源而导致内存不足。然后,大部分工作[46–50]是探索如何通过对每层GCNs的节点进行采样来降低培训成本。然而,这些方法在准确性、可扩展性和训练复杂性方面仍然面临挑战[51,52]。因此,基于子图的方法[51,53,54]被设计成适合大型图和深层网络。受他们想法的启发,本研究对原始图的子图进行采样,并针对每个子图运行完整的GCN模型。为了确保这些子图保留大部分原始边缘,同时仍然呈现有意义的拓扑,我们执行了基于边缘的采样策略,并添加了归一化和方差减少技术。
另一方面,大多数现有的丙二醛预测方法是在平衡数据上训练和测试的,如[55–58]。他们将已知的计量吸入器视为阳性样本,将未知的计量吸入器视为阴性样本,然后对与阳性样本相同数量的阴性样本进行采样,使得阳性样本与阴性样本的比例为1:1。值得注意的是,这些平衡数据的分布不符合计量吸入器的自然分布。虽然许多方法在这些平衡数据上取得了良好的性能,但这并不意味着它们在真实的MDA预测任务上的高性能,因为测试集是不完整的。因此,有必要考虑自然的不平衡数据,尽管不平衡问题仍然是机器学习方法的一个主要挑战[59]。另一方面,现有方法在训练和测试时只考虑新的MiRNA-疾病对(MDPs)的预测,即本研究中的任务对(即Tp)。虽然目前的大多数方法已经对某些疾病进行了案例研究,但仍然不足以说明新的微小核糖核酸和未出现在训练集中的疾病的总体预测性能。因此,本研究同时考虑了上述两类观点,首次提出了关于MDA预测问题的六个实验任务,即分别在平衡和非平衡数据上预测新的MDPs (Tp)、预测新的miRNAs ™和预测新的疾病(Td)。值得注意的是,上述任务中新对象对应的正样本只在测试集中,不在训练集中。
本研究提出了一种新的MDA-GCNFTG方法(图1)用于MDA预测,并在六种不同的预测任务上实现。

该方法主要由两部分组成。首先,我们定义了特征和拓扑图,通过k-最近邻(k-NN)算法[60,61]充分挖掘节点(即miRNA和疾病)特征、网络拓扑(即MDA或miRNA疾病链接)及其组合,为MDA预测任务引入最有用和最深的相关信息。
对于这个图,节点是MDP,节点标签表示MDP是否是MDA,并且基于节点信息在节点和它的k个最近邻居之间构建边。值得注意的是,关于使用MDP作为节点,我们考虑了两个原因:(1)基于相似的微小核糖核酸更可能与相似的疾病相关的假设,反之亦然[62],此外,相似的微小核糖核酸往往具有相似的关联(即标签)。在该图上实施GCN算法将使相似的节点(即相似的微小核糖核酸-疾病对)被聚类。(ii)与异构图相比,同构图更容易学习。然后,在特征图和拓扑图上实现了一种新的基于图采样的GCN算法。
实验结果表明,所提出的MDA-GCNFTG方法在所有六个任务中都取得了满意的结果,在MDA预测问题上优于几种经典的机器学习算法和最新的方法。
此外,本研究还验证了k-神经网络和新的GCN算法在该方法中的有效性。更重要的是,我们分别对微小核糖核酸和疾病进行了两种类型的案例研究。实验结果证明了该方法的有效性和令人满意的性能。
三、相关工作
近年来,已经开发了大量用于丙二醛预测问题的计算方法[63],可分为四类[27],包括基于分数函数的方法、基于多种生物信息的方法、基于复杂网络算法的方法和基于机器学习的方法。基于评分函数的方法[62,64–66]基于训练数据的概率分布或统计分析来定义评分函数,以测量潜在的主成分分析的程度。多种基于生物信息的方法[67–99]考虑了与微小核糖核酸和疾病相关的生物信息学知识,这些知识也可能包括环状核糖核酸、核糖核酸、非环状核糖核酸、药物、蛋白质、微生物等实体。通过上述实体构建的异构图以及它们之间的关系可以为构建微小核糖核酸与疾病之间的关系提供有价值的信息。基于复杂网络算法的方法[100–191]主要基于不同角度的各种疾病和miRNA相似性网络来预测潜在的MDA。基于机器学习的方法[55,192–228]是主成分分析预测领域的重要分支。他们使用机器学习算法来提取有效的特征表示,并解决特定的优化问题,以获得可靠的MDA预测。值得注意的是,以上四种类型的方法并非没有交集。例如,GraRep方法[229]同时采用了多种基于生物信息、基于复杂网络算法和基于机器学习的方法。它建立了一个包含微小核糖核酸、疾病、药物、蛋白质、基因以及它们之间关联的异构图形网络。在嵌入表示的构造中,还考虑了疾病的相似性信息。最后,利用机器学习算法中的随机森林算法对潜在的MDA进行预测。
一些研究试图将GCNs应用于MDA预测问题,它们可以分为以下四类。(I)成对GCNs方法[57,230,231],使用两个GCNs提取微小核糖核酸和疾病嵌入,然后预测计量吸入器。这种方法不考虑MDP之间的连接。(ii)二部图的链接预测方法[56],该方法使用微小核糖核酸和疾病作为节点,计量吸入器作为边,以及基因控制网络来预测潜在的计量吸入器。它将负样本视为参与节点更新的一种边,导致过多的伪邻居造成的过光滑问题。(三)基于完全连通图的GCN方法[58]。由于图过于密集,节点更新后,每个节点的嵌入趋于统一,导致超moothing问题。(4)潘等人提出的研究[232,233]使用多标签以半监督的方式推断疾病相关的微小核糖核酸。然而,这两种方法只实现了丙二醛预测的一部分,而没有考虑对给定疾病预测其相关微小核糖核酸的任务。
四、材料和方法
数据集:
本研究采用人类miRNA疾病数据库(HMDD) v2.0 [234]作为基准数据集。有5430个经实验验证的多药耐药基因,由495个微小核糖核酸和383种疾病组成。在主成分分析预测问题中,已知的主成分分析被视为阳性样本,阴性数据包含所有未知或不存在的主成分分析。本研究中使用的HMDD v2.0于2014年发布。最近的一些研究[55,71,73,172,176,209]使用了2019年发布的HMDD v3.0 [235]的更新数据集。因此,我们还使用了更大的新版本HMDD v3.0来训练和测试我们的方法。HMDD v3.0的统计信息见S1补充表,见http://bib.oxfordjournals.org/.在线提供的补充数据
实验设置和任务:
本研究通过三个实验设置来评估MDA预测问题:(1)任务对(Tp),预测新的MDPs(ii)预测新疾病的任务疾病(Td)和(iii)预测新miRNAs的任务miRNAs ™。相应任务中新对象的标签在训练集中是缺失的,但在测试集中是存在的,用来预测和评估模型性能(如表1所示)。

另一方面,我们在平衡和不平衡数据上评估上述三个实验设置。对于不平衡任务,我们考虑这三个任务中MDAs的整个空间进行更实际的模拟,即使用所有负数据作为负样本参与训练和测试。因此,阳性样本的数量远远低于阴性样本的数量,导致数据不平衡。而且,我们还使用平衡数据来遵循前面的工作,即在训练和测试之前,在与负样本相同的负数据中采样与正样本相同数量的数据。详情如表2所示。

最后,本研究执行了六项任务,涵盖了计量吸入器预测的大多数病例。
五、节点特征构造
本研究采用了基于疾病语义相似性、微小核糖核酸功能相似性和高斯交互分布核相似性的综合特征。特征生成过程如图2所示。

miRNA功能相似矩阵
miRNA功能相似矩阵(MFSM)是基于这样的假设建立的,即具有相似功能的微小核糖核酸更有可能与具有相似表型的疾病相关,反之亦然[236]。相似性信息可以从https://www.cuilab.cn/files/images/cuilab/misim.zip.获得。
疾病语义相似度矩阵
疾病语义相似性可以基于医学主题词描述符[195]来计算,其可以从https://www.ncbi.nlm.nih.gov/.获得。许多研究[56,62,236]使用==有向无环图(DAG)==来生成疾病语义相似性矩阵(DSSM),其中DAG描述了不同疾病的关系。
从两个方面考虑,定义了两种不同的DSM。DSSM1is是基于这样的假设生成的,即如果两种疾病共享其大部分DAG,它们可以被认为更相似。进一步认为,如果疾病出现在更多(或更少)的DAG中,它可能更常见(或更具体)。所以在DAG的同一层,疾病的语义贡献值应该是不同的。这两个DSM来自GAMEDA [56]。
为了获得更合理的DSSM,我们对上述两个数字信号处理器执行元素平均以合成最终的DSSM。
GIP内核相似性
基于相似微小核糖核酸更可能与相似疾病相关的假设[62],可以计算微小核糖核酸(MGSM)和疾病(DGSM)的GIP核相似矩阵。
以建设DGSM为例。
此外,由于规范化,这个内核独立于数据集的大小。MGSM的计算方法与DGSM相似。
集成相似性作为节点特征
考虑到上述MFSM和DSSM中有许多稀疏值,我们融合GIP核相似性MGSM和DGSM分别填充零值。然后,获得整合的微小核糖核酸和疾病相似性矩阵(即免疫球蛋白和免疫球蛋白)。积分方程[62]是

然后,针对所提出的MDA-GCNFTG方法,将IM和ID拼接为特征和拓扑图的节点(即MDP)特征
六、方法
MDA-GCNFTG方法中有两个关键步骤(图1):(1)通过集成的相似性和k-NN算法构造特征和拓扑图,(2)通过图形采样用新的GCN算法预测MDA。
先期工作
定义一个图G (V,E,X),其中V为节点集,E为边集,X为节点特征矩阵。该图描述了具有属性的节点之间的关系。将GCN算法应用于图数据以预测每个节点的类别是节点分类任务。为了对节点进行分类,GCNs使用节点本身的特征以及相邻节点和边缘的信息进行消息传递,这可以多次执行,以聚合来自更广泛的相邻节点的信息来更新节点特征。GCN是一个神经网络层,它从l层到l+1层的传播模式是

其中,A为邻接矩阵,I为单位矩阵,H(l)为lth层的特征,W(l)为lth层的权重,σ为非线性激活函数。对于输入层,H为x。
对于节点分类问题,假设我们构造两层GCN,激活函数分别采用ReLU和softmax,那么总体正向传播公式为

最后,计算所有标记节点的交叉熵损失函数。
通过k-神经网络算法构造特征和拓扑图
有一项研究表明[237]GCNs集成网络拓扑和节点特征以提取与任务最相关的信息的能力并不理想,这可能会严重阻碍分类任务的执行。此外,特征和从拓扑结构推断的信息之间的相似性是相互补充的,并且融合它们可以为分类任务获得更深层次的相关信息[237]。此外,图形数据和任务之间的相关性往往是复杂和不可知的,因此在实际应用中自适应能力也很重要。
受上述研究的启发,本研究提出了一种自适应的图构建方法,该方法能够自适应地将节点特征和拓扑信息传播到特征空间。为了充分捕捉特征空间中的结构信息,基于节点特征和微小核糖核酸与疾病之间的拓扑关系,通过kNN算法生成特征和拓扑图。为了实现这一点,在图中使用MDP作为节点,节点特征是微小核糖核酸和疾病的综合相似性,每个节点的标签代表微小核糖核酸和疾病之间是否存在关联。与现有的基于GCN的主成分分析预测方法相比,该图构建策略不仅考虑了主成分分析之间的关联性,而且实现了异质性的有效融合。最后,生成一个同构图来完成MDA预测问题的节点分类任务。
生成特征和拓扑图的过程是拟合一个k-NN分类器,预测每个节点的标签,并使用k个最近的正确分类的节点作为该节点的邻居。显然,k-NN算法的结果很大程度上取决于K的选择。因此,我们调整K参数(即在1、3、5、7、10或15的值)以研究邻居数量对丙二醛预测的影响。另一方面,这种k-NN算法是对所有数据进行的。为了保证测试数据在训练阶段不会泄露,我们将属于测试数据的节点标签设置为0。
基于图形采样和归一化的遗传神经网络
在本研究中,将一种新的GCN算法应用于MDA预测任务,并且在算法1中说明了整个训练算法。
该算法与传统的GCN算法在小批量构建上有所不同;它基于图形采样。其思想是先对训练图的多个子图进行采样,然后在每个子图上构造完整的GCN。这样,当在GCN层中传播时,可以从子图中获得精确的节点嵌入,并且采样的节点可以相互支持,而无需从批外收集信息。自然,该算法解决了传统GCN算法通常遇到的邻居爆炸的困境。为了保证训练的准确性,需要一个合适的图形采样器。首先,我们认为对彼此影响较大的节点应该在同一个子图中采样,因此本研究使用了基于拓扑的边缘采样器。但是这种影响驱动的抽样想法会引入偏差。为了消除这种偏差,该算法在聚集节点信息和计算小批量损失时,引入节点和边的采样概率进行归一化。
当定义一个边缘采样器时,要点是具有不可忽略概率的边缘应该被采样。另一方面,它还考虑到应该减少节点在完整的全局控制网中的聚集的方差。据此,定义最佳边缘采样概率(见算法1第7行)。公式表明,如果两个连通节点u和v的邻居非常少(即相互影响很大),那么边概率p(u,v)=p(v,u)会很高。
性能赋值
为了便于与其他方法进行比较,我们遵循了以前的研究[56,62,152,156,195,197,229],并对每项任务进行了5倍交叉验证。我们还开展了全球和本地免交一次的简历(详情见补充部分2,见在线补充数据,网址为http://bib.oxfordjournals.org/)。
对于每个任务的每个折叠,将计算以下指标:

在预测结果中,TP为真阳性,FP为假阳性,FN为假阴性,TN为真阴性。
此外,还计算了精度-召回率曲线(AUPR)和AUC下的面积。
值得注意的是,由于平衡数据和不平衡数据的本质区别,在进行绩效评估时,不同指标的重要性也是不同的。在平衡任务中,以上六个指标都很重要。值得注意的是,查全率和查准率通常是一对矛盾的性能指标,所以F1-score常常用来表征它们的综合性能。因此,在下面的讨论中,性能评估指标的平均值是根据准确性、F1-分数、AUC和AUPR来计算的,用于平衡任务。然而,在不平衡的任务中,准确性意义不大,AUPR可以提供比AUC更好的性能估计,因为它将更严厉地惩罚误报。因此,以下讨论中的平均绩效评估指标是根据F1分数和AUPR计算的。
假设检验
在比较一组任务上的多个算法时ˇ sar[238]推荐Friedman秩检验[239240],它使用秩来实现非参数检验,以验证多个总体分布之间是否存在显著差异。
在本研究中,无效假设是不同方法之间没有差异。假设检验的统计结果(即弗里德曼检验的P值)用于基于显著性水平α确定是否拒绝零假设。如果零假设被拒绝,也就是说,至少两种方法之间的差异在统计上是显著的,我们将随后成对地比较每两种方法。在弗里德曼检验的成对比较分析中,我们使用了Bonferroni调整后的P值来考虑多重比较中的ⅰ型误差扩展问题,因此精度优于使用原始P值。最后,该方法可以指示不同方法之间是否存在显著差异。
七、结果和讨论
k-NN在MDA-GCNFTG方法中的作用
为了验证MDA-GCNFTG方法中k-NN算法的有效性,我们首先比较了1-NN和自循环的边构建方法,因为1-NN类似于自循环,它们都只为图中的每个节点创建一条边。自循环建立了从一个节点到节点本身的边,图中每个节点都没有邻居节点,因此无法进行有效的节点更新。自循环策略作为MDAGCNFTG中边缘构建方法的基线,代表了所提出的MDA-GCNFTG方法的最低性能。1-NN在每个节点和它的一个邻居之间建立一条边。虽然可以执行有效的节点更新,但是图中节点的度太低(只有1个),导致图利用率低。由此可见,1-NN在MDA-GCNFTG中的性能也较低。比较结果如图3所示。
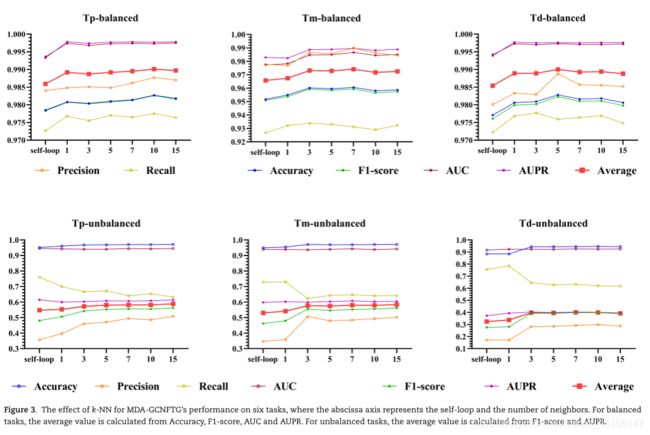
可以看出,1-NN在6项任务上优于自循环法,尤其是在平衡任务上。这证明了通过k-NN算法引入节点间的链接可以提高MDA任务的性能。
然后,我们通过比较k-NN算法中不同数量的最近邻(即k)来阐明模型的鲁棒性。为此,k的值包括1、3、5、7、10和15。
结果表明(图3),对于每个任务,不同k的预测性能大致相等。这表明,所提出的MDA-GCNFTG对k不是很敏感,并且它对边缘构建步骤的鲁棒性得到了证明,这将避免大量的参数调整工作。而且MDAGCNFTG对于不同的任务也有通用性,可以移植到其他的MDA应用中。在HMDD v3.0上进行的实验也证实了上述观点。
此外,我们还从统计学的角度证明了上述观点。每个算法的弗里德曼测试数据都是不同任务的性能评估指标。根据弗里德曼检验计算出的每种方法的平均秩,自循环法排在最后(即平均秩最低),其次是1-NN,说明这两种方法确实不如MDAGCNFTG中的其他k-NN方法。通过对弗里德曼检验的成对比较,自循环与所有六种k-NN方法有显著差异。其用1-NN调整的Bonferroni P值为0.018,用其他5种k-NN方法调整的Bonferroni P值小于0.001。该结果证明了自环和1-神经网络的相似的边缘构建策略都导致相似的较低性能。此外,1-神经网络与5-神经网络和7-神经网络之间的Bonferroni调整后的Pvalues分别为0.025和0.005,这表明它们之间的差异也具有统计学意义。
以上实验表明,k-NN算法能够自适应地提取不同任务的最相关信息,提高分类性能。
MDA-GCNFTG在不同MDA预测任务中的表现
表3显示了所提出的MDA-GCNFTG方法对六个任务的性能,其中每个任务使用从最佳k值建立边缘后获得的预测模型。

结果表明,MDA-GCNFTG在平衡任务上表现出了非凡的预测能力,大部分指标达到了0.98。在不平衡任务上,MDA-GCNFTG看似性能不高,但后面几节的讨论会显示出它的优越性.
另一方面,这是MDA预测问题第一次涉及到除了Tp平衡任务以外的任务,并且都表现出了令人满意的性能。值得注意的是Tm和Td任务比Tp更难。因为他们的目标是预测新的miRNAs和新的疾病,也就是预测训练集中从未出现过的对象。
此外,我们在HMDD v3.0上执行了三项平衡任务,结果可参见S2-S4补充表,参见http://bib.oxfordjournals.org/.在线提供的补充数据。为了遵循之前的研究[207,210,241],我们还在传统的Tp平衡任务上执行了全局和局部遗漏CV。满意的结果可以在补充部分2中看到,参见http://bib.oxfordjournals.org/.在线提供的补充数据
新GCN算法在MDA-GCNFTG方法中的作用
为了证明本文提出的新GCN算法在MDA预测任务中的有效性,我们将其与传统的GCN算法进行了比较。在实现GCN算法时,其实验条件与MDA-GCNFTG方法完全相同,包括5重CV、数据分割、随机种子、边或图构造等。
结果显示(图4),
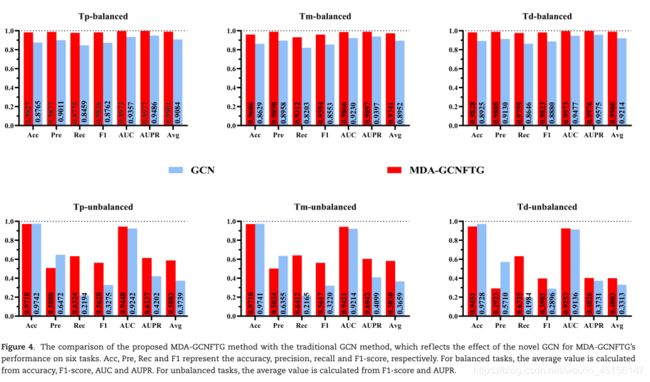
所提出的MDA-GCNFTG方法在所有六个任务上都比GCN具有更高的性能。根据平均性能评价指标,它在两个困难任务(不平衡数据上的Tp和Tm任务)上的优势更加显著。这不仅说明了这种新的GCN算法在MDA预测任务上的有效性,而且证明了本研究提出的新的GCN算法和MDA-GCNFTG方法相对于传统的GCN方法的优越性。在HMDD v3.0上进行的实验也证实了上述观点(见补充表S5-S7,见网上可获得的补充数据,网址为http://bib.oxfordjournals.org/)。
我们还比较了在内存为10,018 MB的英伟达GeForce RTX 3080上提出的新GCN算法和传统GCN算法的时间和内存差异。除了历元设为1外,本实验的所有实验条件同上。

结果如表4所示。对于平衡任务,两种方法的内存消耗非常接近。但在运行速度上,MDA-GCNFTG优势明显,尤其是在Tp和Td任务上,比GCNs快一倍。在不平衡任务上,gcn由于内存不足无法在Tp和Tm任务上运行,在CPU上运行非常慢。
MDA-GCNFTG可以在所有三个不平衡的任务上运行,并且只使用大约7000 MB的内存。
在Td任务中,MDA-GCNFTG不仅比GCN方法消耗的内存少,而且运行速度也比GCN方法快得多,即它们在一个历元下的运行时间分别为14秒和65秒。
MDA-GCNFTG与经典机器学习模型的比较
为了说明本研究提出的GCNFTG模型在丙二醛预测中的优势,我们还将其与一些经典的机器学习算法进行了比较,包括基于深度学习的深度神经网络(DNN)、射频、极随机树(ERTs)、决策树(DTs)和高斯朴素贝叶斯(GNBs)。上述六个任务模型的结果如图5所示。

结果表明,所提出的GCNFTG模型优于其他机器学习模型,尤其是在平衡任务方面。对于不平衡任务,虽然MDA-GCNFTG的召回率比其他模型低,但要认识到精度和召回率是相互矛盾的度量标准。所以两者结合的F1-评分值得关注,显示了MDA-GCNFTG相对于其他方法的优越性。而且,F1和AUPR的平均得分再次证明了这一点。为了充分检验该方法相对于其他经典机器学习算法的优越性,我们进行了弗里德曼检验。虽然不同的绩效评估指标对不平衡的任务有不同的重要性,但测试仍然在所有的绩效评估指标上进行。结果表明,MDA-GCNFTG方法的平均排名最大,即排名第一。此外,MDA-GCNFTG方法优于DT、GNB或DNN算法,显著性水平为0.001,优于ERT算法,显著性水平为0.05。
此外,文中还讨论了MDA-GCNFTG的优越性,通过在HMDD v3.0上进行的实验进行确认(见补充表S5-S7,见补充数据,可在线获取)(http://bib.oxfordjournals.org/)
与最先进方法的比较
为了进一步证明所提出的MDAGCNFTG方法的优越性,我们将其与2020年后发表的三种最先进的方法进行了比较,包括GAMEDA [56]、GBDT -LR [206]和DMA [241]。请注意,以下所有实验都是在相同的实验条件下进行的,包括5倍CV、随机种子和数据分区策略。我们首先验证了MDA-GCNFTG方法在传统任务Tp(即平衡任务Tp)上的优越性,结果表明,MDA-GCNFTG在几乎所有性能评估指标上都优于三种现有方法(图6)。
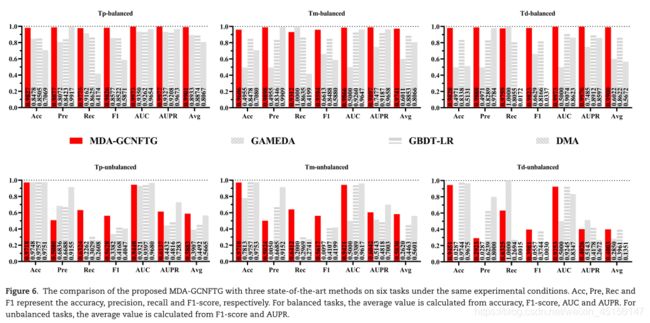
随后,我们修改了GAMEDA、GBDT -LR和DMA的代码以适应本研究中提出的其他五个任务,并在相同的实验条件下将其与提出的MDA-GCNFTG方法进行了比较。结果如图6所示。
对于平衡任务,虽然MDA-GCNFTG的召回率比GAMEDA略低,精度比DMA略低,但总体来说,其性能明显优于这些方法。特别是查全率和查准率通常是一对矛盾的性能指标,发现MDA-GCNFTG的查准率比GAMEDA高很多,而MDA-GCNFTG的查全率比DMA高很多。因此,必须考虑召回率和准确率的综合性能指标,即F1-score和AUPR,并表明MDA-GCNFTG在这两个性能指标上高于GAMEDA和DMA。进一步,我们探究了GAMEDA之所以取得如此高的召回率(即1)的原因,发现它预测所有样本都是阳性样本。而GAMEDA在这两个任务上的AUC都是0.5,也就是说GAMEDA执行的是随机分类,所以其他的性能指标似乎没有意义。不平衡任务中也出现了类似的现象;因此,f1评分和AUPR被认为是计算绩效评估指标的平均值。并且所提出的MDAGCNFTG对于不平衡任务也取得了较好的性能。
图6显示了MDA-GCNFTG相对于最先进的方法的显著优势,也证实了Tm和Td任务比Tp任务更困难的观点,同时证明了所提出的MDA-GCNFTG方法的鲁棒性;也就是说,在Tm、Td和不平衡任务上取得了令人满意的结果。此外,在HMDD v3.0上进行的实验也证实了所提出的MDA-GCNFTG的优越性(参见补充表S5-S7,参见http://bib.oxfordjournals.org/在线提供的补充数据)
八、案例研究
为了进一步验证该方法在丙二醛预测问题上的性能,本研究分别对疾病和微小核糖核酸进行了两种类型的案例研究。这是第一次在丙二醛预测领域进行微小核糖核酸的案例研究。另一方面,我们讨论两种类型数据的案例研究结果。第一个数据是本研究中使用的HMDD v2.0数据库。然而,该数据库是在2014年提出的,7年间发现了许多新的计量吸入器。因此,我们集成了HMDD v3.2 [235]、miR2Disease [242]和dbDEMC [243]数据库作为第二个数据。值得注意的是,第二数据是基于第一数据的更新和扩展。
对于疾病的案例研究,我们选择了肺肿瘤和乳腺肿瘤。肺癌是最常见的致死性癌症,发病率高。虽然新的药物和治疗方法正在开发中,但其出现晚、预后差和治愈率低仍导致其高死亡率。许多研究[244–246]表明,一些微小核糖核酸可以用作肺癌的生物标志物。乳腺癌是女性最常见的癌症之一,早期发现和治疗可以改善患者的预后[247]。然而,其复杂的临床行为和多样的组织病理学模式构成了巨大的挑战[247]。证据[247]表明,部分微小核糖核酸与乳腺癌有密切关系,因此相关微小核糖核酸可作为检测和预防乳腺癌的生物标志物。
通过对miRNA的广泛研究,已经确定let-7 miRNA家族与hsa-mir-1相关
多种人类疾病[248–250]。Hsa-let-7a可诱导异常表达的疾病[251–253]。
Hsa-let-7b是各种疾病表观遗传机制的重要靶点[253–256]。最近的研究还报道了hsa-mir-1与各种复杂人类疾病之间的联系[257–259],并发现hsa-mir-1在结直肠癌中频繁甲基化,并认为hsa-mir-1通过控制上皮转化因子的表达而发挥肿瘤抑制作用[260,261]。
表5列出了五个案例研究的结果和绩效。
很明显,对整合数据进行的案例研究令人信服,并且都显示出令人满意的结果,证明所提出的MDA-GCNFTG方法能够预测新的微小核糖核酸和新的疾病的未发现的潜在MDA。两类数据上阳性样本数与TP的差异也印证了这一观点,也反映出本研究中所提出的MDA-GCNFTG方法的性能被严重低估。
九、结论
miRNA已被证明与许多复杂的人类疾病密切相关。因此,预测潜在的计量吸入器对理解、预防和治疗疾病至关重要。本研究设计了一种新的图构造策略,利用k-NN算法和基于图抽样技术的GCN模型进行MDA预测,即MDA-GCNFTG方法。此外,与其他仅基于平衡数据预测新MDA的研究相比,本研究提出了两种新的预测新微小核糖核酸和预测新疾病的实验设置,上述三种实验设置将分别在平衡和非平衡数据上进行。结果表明,该方法在6个任务上都取得了令人满意的结果,优于几种经典的机器学习算法和最先进的主成分分析预测方法。此外,我们还对微小核糖核酸和疾病进行了案例研究,这证实了我们方法的有效性。未来,我们希望整合其他生物信息,并将数据预处理技术应用于不平衡数据,以获得更好的结果。此外,大多数关于主成分分析预测的研究(包括本研究)在整个数据集上使用了从已知主成分分析中导出的基于相似性的度量,这导致了当前研究的过度乐观的性能评估。在下一步中,我们将尝试开发一种更合适的特征表示方法。例如,在划分训练集和测试集后,使用测试样本掩蔽方法首先计算训练集样本之间的相似度,然后使用k-NN算法构造测试集样本之间的相似度。

