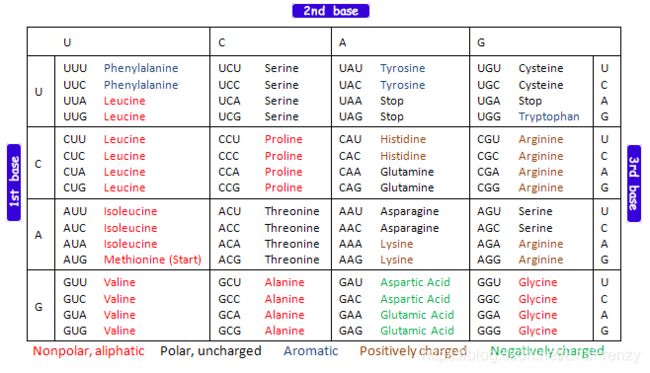
一、什么是宏函数?通过宏定义的函数是宏函数。如下,编译器在预处理阶段会将Add(x,y)替换为((x)*(y))#defineAdd(x,y)((x)*(y))#defineAdd(x,y)((x)*(y))intmain(){inta=10;intb=20;intd=10;intc=Add(a+d,b)*2;cout<
C语言如何定义宏函数?
小九格物
c语言
在C语言中,宏函数是通过预处理器定义的,它在编译之前替换代码中的宏调用。宏函数可以模拟函数的行为,但它们不是真正的函数,因为它们在编译时不会进行类型检查,也不会分配存储空间。宏函数的定义通常使用#define指令,后面跟着宏的名称和参数列表,以及宏展开后的代码。宏函数的定义方式:1.基本宏函数:这是最简单的宏函数形式,它直接定义一个表达式。#defineSQUARE(x)((x)*(x))2.带参
微服务下功能权限与数据权限的设计与实现
nbsaas-boot
微服务java架构
在微服务架构下,系统的功能权限和数据权限控制显得尤为重要。随着系统规模的扩大和微服务数量的增加,如何保证不同用户和服务之间的访问权限准确、细粒度地控制,成为设计安全策略的关键。本文将讨论如何在微服务体系中设计和实现功能权限与数据权限控制。1.功能权限与数据权限的定义功能权限:指用户或系统角色对特定功能的访问权限。通常是某个用户角色能否执行某个操作,比如查看订单、创建订单、修改用户资料等。数据权限:
理解Gunicorn:Python WSGI服务器的基石
范范0825
ipythonlinux运维
理解Gunicorn:PythonWSGI服务器的基石介绍Gunicorn,全称GreenUnicorn,是一个为PythonWSGI(WebServerGatewayInterface)应用设计的高效、轻量级HTTP服务器。作为PythonWeb应用部署的常用工具,Gunicorn以其高性能和易用性著称。本文将介绍Gunicorn的基本概念、安装和配置,帮助初学者快速上手。1.什么是Gunico
小丽成长记(四十三)
玲玲54321
小丽发现,即使她好不容易调整好自己的心态下一秒总会有不确定的伤脑筋的事出现,一个接一个的问题,人生就没有停下的时候,小问题不断出现。不过她今天看的书,她接受了人生就是不确定的,厉害的人就是不断创造确定性,在Ta的领域比别人多的确定性就能让自己脱颖而出,显示价值从而获得的比别人多的利益。正是这样的原因,因为从前修炼自己太少,使得她现在在人生道路上打怪起来困难重重,她似乎永远摆脱不了那种无力感,有种习
2021年12月19日,春蕾教育集团团建活动感受——黄晓丹
黄错错加油
感受:1.从陌生到熟悉的过程。游戏环节让我们在轻松的氛围中得到了锻炼,也增长了不少知识。2.游戏过程中,我们贡献的是个人力量,展现的是团队的力量。它磨合的往往不止是工作的熟悉,更是观念上契合度的贴近。3.这和工作是一样的道理。在各自的岗位上,每个人摆正自己的位置、各司其职充分发挥才能,并团结一致劲往一处使,才能实现最大的成功。新知:1.团队精神需要不断地创新。过去,人们把创新看作是冒风险,现在人们
c++ 的iostream 和 c++的stdio的区别和联系
黄卷青灯77
c++算法开发语言iostreamstdio
在C++中,iostream和C语言的stdio.h都是用于处理输入输出的库,但它们在设计、用法和功能上有许多不同。以下是两者的区别和联系:区别1.编程风格iostream(C++风格):C++标准库中的输入输出流类库,支持面向对象的输入输出操作。典型用法是cin(输入)和cout(输出),使用>操作符来处理数据。更加类型安全,支持用户自定义类型的输入输出。#includeintmain(){in
爬山后遗症
璃绛
爬山,攀登,一步一步走向制高点,是一种挑战。成功抵达是一种无法言语的快乐,在山顶吹吹风,看看风景,这是从未有过的体验。然而,爬山一时爽,下山腿打颤,颠簸的路,一路向下走,腿部力量不够,走起来抖到不行,停不下来了!第二天必定腿疼,浑身酸痛,坐立难安!
Long类型前后端数据不一致
igotyback
前端
响应给前端的数据浏览器控制台中response中看到的Long类型的数据是正常的到前端数据不一致前后端数据类型不匹配是一个常见问题,尤其是当后端使用Java的Long类型(64位)与前端JavaScript的Number类型(最大安全整数为2^53-1,即16位)进行数据交互时,很容易出现精度丢失的问题。这是因为JavaScript中的Number类型无法安全地表示超过16位的整数。为了解决这个问
swagger访问路径
igotyback
swagger
Swagger2.x版本访问地址:http://{ip}:{port}/{context-path}/swagger-ui.html{ip}是你的服务器IP地址。{port}是你的应用服务端口,通常为8080。{context-path}是你的应用上下文路径,如果应用部署在根路径下,则为空。Swagger3.x版本对于Swagger3.x版本(也称为OpenAPI3)访问地址:http://{ip
mysql禁用远程登录
igotyback
mysql
去mysql库中的user表里,将host都改成localhost之后刷新权限FLUSHPRIVILEGES;
如何在 Fork 的 GitHub 项目中保留自己的修改并同步上游更新?github_fork_update
iBaoxing
github
如何在Fork的GitHub项目中保留自己的修改并同步上游更新?在GitHub上Fork了一个项目后,你可能会对项目进行一些修改,同时原作者也在不断更新。如果想要在保留自己修改的基础上,同步原作者的最新更新,很多人会不知所措。本文将详细讲解如何在不丢失自己改动的情况下,将上游仓库的更新合并到自己的仓库中。问题描述假设你在GitHub上Fork了一个项目,并基于该项目做了一些修改,随后你发现原作者对
Linux下QT开发的动态库界面弹出操作(SDL2)
13jjyao
QT类qt开发语言sdl2linux
需求:操作系统为linux,开发框架为qt,做成需带界面的qt动态库,调用方为java等非qt程序难点:调用方为java等非qt程序,也就是说调用方肯定不带QApplication::exec(),缺少了这个,QTimer等事件和QT创建的窗口将不能弹出(包括opencv也是不能弹出);这与qt调用本身qt库是有本质的区别的思路:1.调用方缺QApplication::exec(),那么我们在接口
30天风格练习-DAY2
黄希夷
Day2(重义)在一个周日/一周的最后一天,我来到位于市中心/市区繁华地带的一家购物中心/商场,中心内人很多/熙熙攘攘。我注意到/看见一个独行/孤身一人的年轻女孩/,留着一头引人注目/长过腰际的头发,上身穿一件暗红色/比正红色更深的衣服/穿在身体上的东西。走下扶梯的时候,她摔倒了/跌向地面,在她正要站起来/让身体离开地面的时候,过长/超过一般人长度的头发被支撑身体/躯干的手掌压/按在下面,她赶紧用
店群合一模式下的社区团购新发展——结合链动 2+1 模式、AI 智能名片与 S2B2C 商城小程序源码
说私域
人工智能小程序
摘要:本文探讨了店群合一的社区团购平台在当今商业环境中的重要性和优势。通过分析店群合一模式如何将互联网社群与线下终端紧密结合,阐述了链动2+1模式、AI智能名片和S2B2C商城小程序源码在这一模式中的应用价值。这些创新元素的结合为社区团购带来了新的机遇,提升了用户信任感、拓展了营销渠道,并实现了线上线下的完美融合。一、引言随着互联网技术的不断发展,社区团购作为一种新兴的商业模式,在满足消费者日常需
我校举行新老教师师徒结对仪式暨名师专业工作室工作交流活动
李蕾1229
为促进我校教师专业发展,发挥骨干教师的引领带头作用,11月6日下午,我校举行新老教师师徒结对仪式暨名师专业工作室工作交流活动。图片发自App会议由教师发展处李蕾主任主持,首先,由范校长宣读新老教师结对名单及双方承担职责。随后,两位新调入教师陈玉萍、莫正杰分别和他们的师傅鲍元美、刘召彬老师签订了师徒结对协议书。图片发自App图片发自App师徒拥抱、握手。有了师傅就有了目标有了方向,相信两位新教师在师
向内而求
陈陈_19b4
10月27日,阴。阅读书目:《次第花开》。作者:希阿荣博堪布,是当今藏传佛家宁玛派最伟大的上师法王,如意宝晋美彭措仁波切颇具影响力的弟子之一。多年以来,赴海内外各地弘扬佛法,以正式授课、现场开示、发表文章等多种方法指导佛学弟子修行佛法。代表作《寂静之道》、《生命这出戏》、《透过佛法看世界》自出版以来一直是佛教类书籍中的畅销书。图片发自App金句:1.佛陀说,一切痛苦的根源在于我们长期以来对自身及外
消息中间件有哪些常见类型
xmh-sxh-1314
java
消息中间件根据其设计理念和用途,可以大致分为以下几种常见类型:点对点消息队列(Point-to-PointMessagingQueues):在这种模型中,消息被发送到特定的队列中,消费者从队列中取出并处理消息。队列中的消息只能被一个消费者消费,消费后即被删除。常见的实现包括IBM的MQSeries、RabbitMQ的部分使用场景等。适用于任务分发、负载均衡等场景。发布/订阅消息模型(Pub/Sub
ArcGIS栅格计算器常见公式(赋值、0和空值的转换、补充栅格空值)
研学随笔
arcgis经验分享
我们在使用ArcGIS时通常经常用到栅格计算器,今天主要给大家介绍我日常中经常用到的几个公式,供大家参考学习。将特定值(-9999)赋值为0,例如-9999.Con("raster"==-9999,0,"raster")2.给空值赋予特定的值(如0)Con(IsNull("raster"),0,"raster")3.将特定的栅格值(如1)赋值为空值,其他保留原值SetNull("raster"==
那个抄袭的大张伟
猫小努
最近一直在追《即刻电音》这个综艺,除了觉得出场节目的音乐制作人有意思之外,也觉得有两个导师挺有趣的(另外一个就忽略了吧)。孙艺兴在上一篇文章里面已经说过了,那么这篇就说说我们的大老师,大张伟吧。其实在节目刚开始大张伟出来的时候,我以为他是属于导师里面来活跃气氛负责搞笑的,毕竟孙艺兴属于卖萌卖傻卖老实的,尚雯婕一般负责装逼耍狠的,而大张伟一贯以来上综艺的形象基本上都是蹦蹦跳跳带动气氛的。谁知道,两期
三大师传
beca酱
巴尔扎克的作品被誉为“法国社会的一面镜子”。文学大师维克多·雨果对巴尔扎克的评价是:“在最伟大的人物中间,巴尔扎克是名列前茅者;在最优秀的人物中间,巴尔扎克是佼佼者之一。”一个原本寂寂无名的小人物,从地中海的某个海岛上,只身一人来到巴黎,没有朋友,也没有名望。作为一个一文不名的外乡人,凭着赤手空拳赢得了巴黎,征服了整个法兰西,并且赢得了世界。这个人就是十九世纪法国伟大的军事家、政治家,法兰西第一帝
2018-07-23-催眠日作业-#不一样的31天#-66小鹿
小鹿_33
预言日:人总是在逃避命运的路上,与之不期而遇。心理学上有个著名的名词,叫做自证预言;经济学上也有一个很著名的定律叫做,墨菲定律;在灵修派上,还有一个很著名的法则,叫做吸引力法则。这3个领域的词,虽然看起来不太一样,但是他们都在告诉人们一个现象:你越担心什么,就越有可能会发生什么。同样的道理,你越想得到什么,就应该要积极地去创造什么。无论是自证预言,墨菲定律还是吸引力法则,对人都有正反2个维度的影响
Java实现的基于模板的网页结构化信息精准抽取组件:HtmlExtractor
yangshangchuan
信息抽取HtmlExtractor精准抽取信息采集
HtmlExtractor是一个Java实现的基于模板的网页结构化信息精准抽取组件,本身并不包含爬虫功能,但可被爬虫或其他程序调用以便更精准地对网页结构化信息进行抽取。
HtmlExtractor是为大规模分布式环境设计的,采用主从架构,主节点负责维护抽取规则,从节点向主节点请求抽取规则,当抽取规则发生变化,主节点主动通知从节点,从而能实现抽取规则变化之后的实时动态生效。
如
java编程思想 -- 多态
百合不是茶
java多态详解
一: 向上转型和向下转型
面向对象中的转型只会发生在有继承关系的子类和父类中(接口的实现也包括在这里)。父类:人 子类:男人向上转型: Person p = new Man() ; //向上转型不需要强制类型转化向下转型: Man man =
[自动数据处理]稳扎稳打,逐步形成自有ADP系统体系
comsci
dp
对于国内的IT行业来讲,虽然我们已经有了"两弹一星",在局部领域形成了自己独有的技术特征,并初步摆脱了国外的控制...但是前面的路还很长....
首先是我们的自动数据处理系统还无法处理很多高级工程...中等规模的拓扑分析系统也没有完成,更加复杂的
storm 自定义 日志文件
商人shang
stormclusterlogback
Storm中的日志级级别默认为INFO,并且,日志文件是根据worker号来进行区分的,这样,同一个log文件中的信息不一定是一个业务的,这样就会有以下两个需求出现:
1. 想要进行一些调试信息的输出
2. 调试信息或者业务日志信息想要输出到一些固定的文件中
不要怕,不要烦恼,其实Storm已经提供了这样的支持,可以通过自定义logback 下的 cluster.xml 来输
Extjs3 SpringMVC使用 @RequestBody 标签问题记录
21jhf
springMVC使用 @RequestBody(required = false) UserVO userInfo
传递json对象数据,往往会出现http 415,400,500等错误,总结一下需要使用ajax提交json数据才行,ajax提交使用proxy,参数为jsonData,不能为params;另外,需要设置Content-type属性为json,代码如下:
(由于使用了父类aaa
一些排错方法
文强chu
方法
1、java.lang.IllegalStateException: Class invariant violation
at org.apache.log4j.LogManager.getLoggerRepository(LogManager.java:199)at org.apache.log4j.LogManager.getLogger(LogManager.java:228)
at o
Swing中文件恢复我觉得很难
小桔子
swing
我那个草了!老大怎么回事,怎么做项目评估的?只会说相信你可以做的,试一下,有的是时间!
用java开发一个图文处理工具,类似word,任意位置插入、拖动、删除图片以及文本等。文本框、流程图等,数据保存数据库,其余可保存pdf格式。ok,姐姐千辛万苦,
php 文件操作
aichenglong
PHP读取文件写入文件
1 写入文件
@$fp=fopen("$DOCUMENT_ROOT/order.txt", "ab");
if(!$fp){
echo "open file error" ;
exit;
}
$outputstring="date:"." \t tire:".$tire."
MySQL的btree索引和hash索引的区别
AILIKES
数据结构mysql算法
Hash 索引结构的特殊性,其 检索效率非常高,索引的检索可以一次定位,不像B-Tree 索引需要从根节点到枝节点,最后才能访问到页节点这样多次的IO访问,所以 Hash 索引的查询效率要远高于 B-Tree 索引。
可能很多人又有疑问了,既然 Hash 索引的效率要比 B-Tree 高很多,为什么大家不都用 Hash 索引而还要使用 B-Tree 索引呢
JAVA的抽象--- 接口 --实现
百合不是茶
抽象 接口 实现接口
//抽象 类 ,方法
//定义一个公共抽象的类 ,并在类中定义一个抽象的方法体
抽象的定义使用abstract
abstract class A 定义一个抽象类 例如:
//定义一个基类
public abstract class A{
//抽象类不能用来实例化,只能用来继承
//
JS变量作用域实例
bijian1013
作用域
<script>
var scope='hello';
function a(){
console.log(scope); //undefined
var scope='world';
console.log(scope); //world
console.log(b);
TDD实践(二)
bijian1013
javaTDD
实践题目:分解质因数
Step1:
单元测试:
package com.bijian.study.factor.test;
import java.util.Arrays;
import junit.framework.Assert;
import org.junit.Before;
import org.junit.Test;
import com.bijian.
[MongoDB学习笔记一]MongoDB主从复制
bit1129
mongodb
MongoDB称为分布式数据库,主要原因是1.基于副本集的数据备份, 2.基于切片的数据扩容。副本集解决数据的读写性能问题,切片解决了MongoDB的数据扩容问题。
事实上,MongoDB提供了主从复制和副本复制两种备份方式,在MongoDB的主从复制和副本复制集群环境中,只有一台作为主服务器,另外一台或者多台服务器作为从服务器。 本文介绍MongoDB的主从复制模式,需要指明
【HBase五】Java API操作HBase
bit1129
hbase
import java.io.IOException;
import org.apache.hadoop.conf.Configuration;
import org.apache.hadoop.hbase.HBaseConfiguration;
import org.apache.hadoop.hbase.HColumnDescriptor;
import org.apache.ha
python调用zabbix api接口实时展示数据
ronin47
zabbix api接口来进行展示。经过思考之后,计划获取如下内容: 1、 获得认证密钥 2、 获取zabbix所有的主机组 3、 获取单个组下的所有主机 4、 获取某个主机下的所有监控项
jsp取得绝对路径
byalias
绝对路径
在JavaWeb开发中,常使用绝对路径的方式来引入JavaScript和CSS文件,这样可以避免因为目录变动导致引入文件找不到的情况,常用的做法如下:
一、使用${pageContext.request.contextPath}
代码” ${pageContext.request.contextPath}”的作用是取出部署的应用程序名,这样不管如何部署,所用路径都是正确的。
Java定时任务调度:用ExecutorService取代Timer
bylijinnan
java
《Java并发编程实战》一书提到的用ExecutorService取代Java Timer有几个理由,我认为其中最重要的理由是:
如果TimerTask抛出未检查的异常,Timer将会产生无法预料的行为。Timer线程并不捕获异常,所以 TimerTask抛出的未检查的异常会终止timer线程。这种情况下,Timer也不会再重新恢复线程的执行了;它错误的认为整个Timer都被取消了。此时,已经被
SQL 优化原则
chicony
sql
一、问题的提出
在应用系统开发初期,由于开发数据库数据比较少,对于查询SQL语句,复杂视图的的编写等体会不出SQL语句各种写法的性能优劣,但是如果将应用系统提交实际应用后,随着数据库中数据的增加,系统的响应速度就成为目前系统需要解决的最主要的问题之一。系统优化中一个很重要的方面就是SQL语句的优化。对于海量数据,劣质SQL语句和优质SQL语句之间的速度差别可以达到上百倍,可见对于一个系统
java 线程弹球小游戏
CrazyMizzz
java游戏
最近java学到线程,于是做了一个线程弹球的小游戏,不过还没完善
这里是提纲
1.线程弹球游戏实现
1.实现界面需要使用哪些API类
JFrame
JPanel
JButton
FlowLayout
Graphics2D
Thread
Color
ActionListener
ActionEvent
MouseListener
Mouse
hadoop jps出现process information unavailable提示解决办法
daizj
hadoopjps
hadoop jps出现process information unavailable提示解决办法
jps时出现如下信息:
3019 -- process information unavailable3053 -- process information unavailable2985 -- process information unavailable2917 --
PHP图片水印缩放类实现
dcj3sjt126com
PHP
<?php
class Image{
private $path;
function __construct($path='./'){
$this->path=rtrim($path,'/').'/';
}
//水印函数,参数:背景图,水印图,位置,前缀,TMD透明度
public function water($b,$l,$pos
IOS控件学习:UILabel常用属性与用法
dcj3sjt126com
iosUILabel
参考网站:
http://shijue.me/show_text/521c396a8ddf876566000007
http://www.tuicool.com/articles/zquENb
http://blog.csdn.net/a451493485/article/details/9454695
http://wiki.eoe.cn/page/iOS_pptl_artile_281
完全手动建立maven骨架
eksliang
javaeclipseWeb
建一个 JAVA 项目 :
mvn archetype:create
-DgroupId=com.demo
-DartifactId=App
[-Dversion=0.0.1-SNAPSHOT]
[-Dpackaging=jar]
建一个 web 项目 :
mvn archetype:create
-DgroupId=com.demo
-DartifactId=web-a
配置清单
gengzg
配置
1、修改grub启动的内核版本
vi /boot/grub/grub.conf
将default 0改为1
拷贝mt7601Usta.ko到/lib文件夹
拷贝RT2870STA.dat到 /etc/Wireless/RT2870STA/文件夹
拷贝wifiscan到bin文件夹,chmod 775 /bin/wifiscan
拷贝wifiget.sh到bin文件夹,chm
Windows端口被占用处理方法
huqiji
windows
以下文章主要以80端口号为例,如果想知道其他的端口号也可以使用该方法..........................1、在windows下如何查看80端口占用情况?是被哪个进程占用?如何终止等. 这里主要是用到windows下的DOS工具,点击"开始"--"运行",输入&
开源ckplayer 网页播放器, 跨平台(html5, mobile),flv, f4v, mp4, rtmp协议. webm, ogg, m3u8 !
天梯梦
mobile
CKplayer,其全称为超酷flv播放器,它是一款用于网页上播放视频的软件,支持的格式有:http协议上的flv,f4v,mp4格式,同时支持rtmp视频流格 式播放,此播放器的特点在于用户可以自己定义播放器的风格,诸如播放/暂停按钮,静音按钮,全屏按钮都是以外部图片接口形式调用,用户根据自己的需要制作 出播放器风格所需要使用的各个按钮图片然后替换掉原始风格里相应的图片就可以制作出自己的风格了,
简单工厂设计模式
hm4123660
java工厂设计模式简单工厂模式
简单工厂模式(Simple Factory Pattern)属于类的创新型模式,又叫静态工厂方法模式。是通过专门定义一个类来负责创建其他类的实例,被创建的实例通常都具有共同的父类。简单工厂模式是由一个工厂对象决定创建出哪一种产品类的实例。简单工厂模式是工厂模式家族中最简单实用的模式,可以理解为是不同工厂模式的一个特殊实现。
maven笔记
zhb8015
maven
跳过测试阶段:
mvn package -DskipTests
临时性跳过测试代码的编译:
mvn package -Dmaven.test.skip=true
maven.test.skip同时控制maven-compiler-plugin和maven-surefire-plugin两个插件的行为,即跳过编译,又跳过测试。
指定测试类
mvn test
非mapreduce生成Hfile,然后导入hbase当中
Stark_Summer
maphbasereduceHfilepath实例
最近一个群友的boss让研究hbase,让hbase的入库速度达到5w+/s,这可愁死了,4台个人电脑组成的集群,多线程入库调了好久,速度也才1w左右,都没有达到理想的那种速度,然后就想到了这种方式,但是网上多是用mapreduce来实现入库,而现在的需求是实时入库,不生成文件了,所以就只能自己用代码实现了,但是网上查了很多资料都没有查到,最后在一个网友的指引下,看了源码,最后找到了生成Hfile
jsp web tomcat 编码问题
王新春
tomcatjsppageEncode
今天配置jsp项目在tomcat上,windows上正常,而linux上显示乱码,最后定位原因为tomcat 的server.xml 文件的配置,添加 URIEncoding 属性:
<Connector port="8080" protocol="HTTP/1.1"
connectionTi