【R语言】——绘制按聚类结果分组的热图 3(保姆级教程)
上一期”【R语言】——聚类热图行列分组信息注释热图2“介绍了R语言pheatmap包绘制分组信息注释热图,本期主要介绍了另一种分组信息注释方式,通过对数据聚类结果的分析,预设数据聚类簇,从而针对这一数据信息,绘制按聚类结果分组的聚类热图。
1 数据准备
数据输入格式(csv格式):
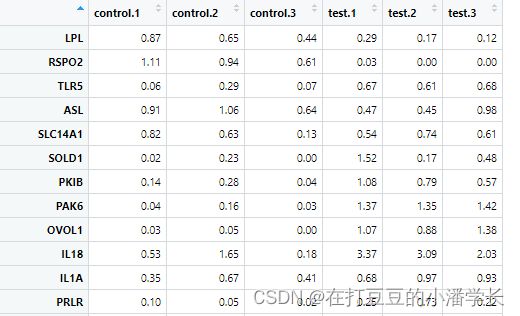
2 R包加载及数据导入
#下载包#
install.packages("pheatmap")
install.packages("RColorBrewer")
#加载包#
library("pheatmap")
library("RColorBrewer")
#加载绘图数据#
data<-read.table(file='C:/Rdata/jc/pheatmap.csv',header=TRUE,row.names= 1,sep=',')
head(data) #查看数据
#data=log2(data[,1:6]+1) #对基因表达量数据处理
#data <- as.matrix(data) #转变为matrix格式矩阵
#head(data)3 按聚类结果分组的热图
3.1 查看数据的分簇数
在绘制由聚类结果分组的热图前,需估计数据的分簇数,从而为后续分簇数选择提供依据。通常,选择类内平方和降低开始趋于平缓的聚类数作为较优聚类数:
#查看数据的分簇数
data <- t(apply(data, 1, scale))
tested_cluster <- 30 #检验的分簇数
wss <- (nrow(data)-1) * sum(apply(data, 2, var))
for (i in 2:tested_cluster) {
wss[i] <- kmeans(data, centers=i,iter.max=100, nstart=25)$tot.withinss
}
plot(1:tested_cluster, wss, type="b", xlab="Number of Clusters", ylab="Within groups sum of squares")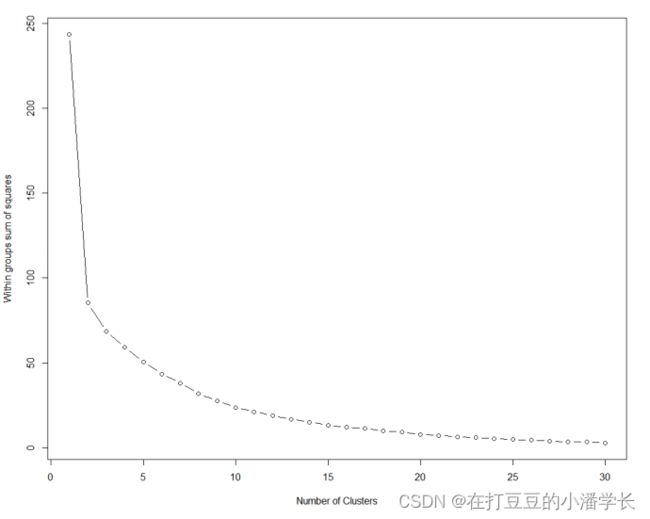
图1 聚类簇图
3.2 生成初始热图
#列信息注释
ann_col = data.frame(Sample=c(rep("control",3),rep("test",3)))#创建分组列
row.names(ann_col) = colnames(data) #这一行必须有,否则会报错:Error in check.length("fill") : 'gpar' element 'fill' must not be length 0
ann_color = list(Sample = c(control="#0089CF", test="#E889BD")) #定义分组颜色#热图绘制
p<-pheatmap(data, scale = "row", #表示进行均一化的方向,值为 “row”, “column” 或者"none"
cluster_rows = T,cluster_cols = T, #cluster_rows表示仅对行聚类,cluster_cols表示仅对列聚类,值为TRUE或FALSE
cutree_rows = NA, cutree_cols = NA, #若进行了行/列聚类,根据行/列聚类数量分隔热图行,cutree_rows=num分割行,cutree_cols=num分割列
treeheight_row = 30, treeheight_col = 30, #若行、列聚类树高度调整
border_color = "grey60", #表示热图每个小的单元格边框的颜色,默认为 "grey60"
cellwidth = 60, cellheight = 7.5, #表示单个单元格的宽度\高度,默认为 “NA”
display_numbers = F, #表示是否在单元格上显示原始数值或按照特殊条件进行区分标记
fontsize_number = 6, #表示热图上显示数字的字体大小
number_format = "%.2f", #表示热图单元格上显示的数据格式,“%.2f” 表示两位小数,“%.1e”表示科学计数法
number_color = "grey30", #表示热图单元格上显示的数据字体颜色
fontsize =10, fontsize_row = 6, fontsize_col = 10, #热图中字体大小、行、列名字体大小
show_rownames = T, show_colnames = T, #表示是否显示行名、列名
main = "Gene标题", #表示热图的标题名字
color = colorRampPalette(c("navy","white","firebrick3"))(100), #表示热图颜色,(100)表示100个等级
angle_col = "45", #表示列标签的角度
gaps_row = NULL, #仅在未进行行聚类时使用,表示在行方向上热图的隔断位置
gaps_col = c(1,2,3,4,5,6), #仅在未进行列聚类时使用,表示在列方向上热图的隔断位置
annotation_row = NA, annotation_col = ann_col, #表示是否对行、列进行注释,默认NA
annotation = NA, annotation_colors = ann_color, #表示行注释及列注释的颜色,默认NA
annotation_legend = TRUE, #表示是否显示注释的图例信息
annotation_names_row = TRUE, annotation_names_col = TRUE) #表示是否显示行、列注释的名称
summary(p)3.3 生成聚类树
利用“tree_row” 和 “tree_col” 提取出对应的行列顺序,进而按预设的分簇数构建分簇数据集,为后续的热图绘制提供分组注释依据:
#提取热图的行方向(基因)的聚类树
clu<- p$tree_row
#clu<- p$tree_row$order
#clu<- p$tree_col$order
#对聚类树进行分簇;
cluster<- factor(cutree(clu,20)) #数值为预设分簇数
cluster
#转成数据框;
cut.df <- data.frame(cluster)
#绘制聚类树;
plot(clu,hang = -1,cex=0.6,axes=FALSE,ann=FALSE)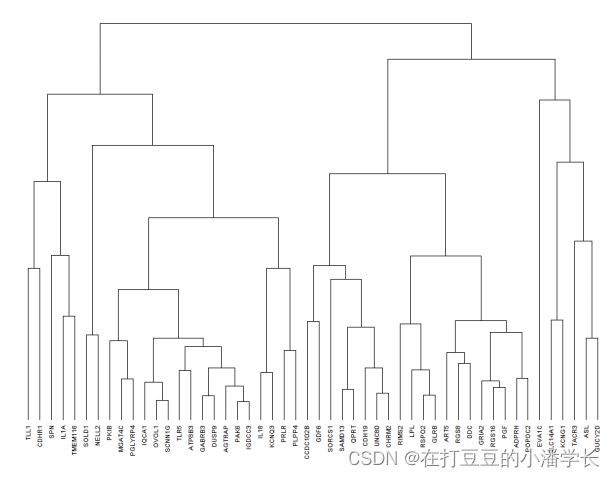
图2 基因聚类树
3.4 按聚类结果绘制热图
利用“annotation_row”将分簇聚类数据结果进行注释,从而绘制聚类热图:
#热图绘制
p<-pheatmap(data, scale = "row", #表示进行均一化的方向,值为 “row”, “column” 或者"none"
cluster_rows = T,cluster_cols = T, #cluster_rows表示仅对行聚类,cluster_cols表示仅对列聚类,值为TRUE或FALSE
cutree_rows = NA, cutree_cols = NA, #若进行了行/列聚类,根据行/列聚类数量分隔热图行,cutree_rows=num分割行,cutree_cols=num分割列
treeheight_row = 30, treeheight_col = 30, #若行、列聚类树高度调整
border_color = "grey60", #表示热图每个小的单元格边框的颜色,默认为 "grey60"
cellwidth = 60, cellheight = 7.5, #表示单个单元格的宽度\高度,默认为 “NA”
display_numbers = F, #表示是否在单元格上显示原始数值或按照特殊条件进行区分标记
fontsize_number = 6, #表示热图上显示数字的字体大小
number_format = "%.2f", #表示热图单元格上显示的数据格式,“%.2f” 表示两位小数,“%.1e”表示科学计数法
number_color = "grey30", #表示热图单元格上显示的数据字体颜色
fontsize =10, fontsize_row = 6, fontsize_col = 10, #热图中字体大小、行、列名字体大小
show_rownames = T, show_colnames = T, #表示是否显示行名、列名
main = "Gene标题", #表示热图的标题名字
color = colorRampPalette(c("navy","white","firebrick3"))(100), #表示热图颜色,(100)表示100个等级
angle_col = "45", #表示列标签的角度
gaps_row = NULL, #仅在未进行行聚类时使用,表示在行方向上热图的隔断位置
gaps_col = c(1,2,3,4,5,6), #仅在未进行列聚类时使用,表示在列方向上热图的隔断位置
annotation_row = cut.df, annotation_col = ann_col, #表示是否对行、列进行注释,默认NA
annotation = NA, annotation_colors = ann_color, #表示行注释及列注释的颜色,默认NA
annotation_legend = TRUE, #表示是否显示注释的图例信息
annotation_names_row = TRUE, annotation_names_col = TRUE) #表示是否显示行、列注释的名称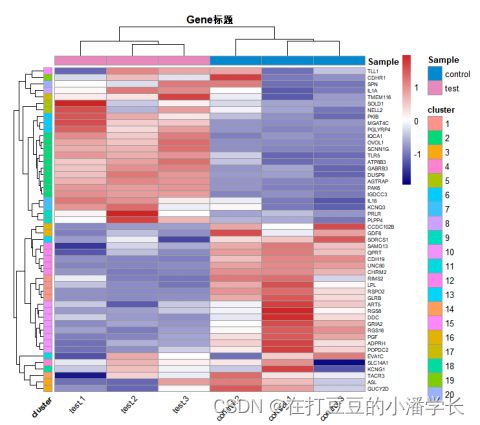
图3 分簇聚类分组热图
好了本次分享就到这里,下一期将分享根据聚类情况绘制热图的分组。

扫码关注公众号,发送”分组热图3“获得完整代码以及演示数据包