Diffusion-weighting in MRI 学习笔记
功能像的学习已经差不多了,对功能连接、图论、机器学习方面的知识感觉已经掌握的差不多了,这段时间准备把DTI像给捡起来学习学习。

图片引用自FSL官网
学习DTI,不能逾越的软件当推FSL,我的学习笔记及部分博文图片也来自于FSL官网教程,仅供学习谈论。
概述
分析DTI像,主要就是集中于构建白质纤维束,要理解白质纤维束的构建过程,追源溯流,最开始的理论起源于分子的布朗运动。
大家都知道,分子处于不断的随机运动中,这种运动被称为布朗运动,也讲作弥散(diffussion)。
Diffusion = thermally-driven random motion
如何描述这种弥散呢?爱因斯坦提出了下述公式:

这种现象在大脑里也是存在的,因为大脑传递信息是通过神经元,而神经元的结构是树突和轴突组成,轴突就是方向性的连接。

在纤维束中,分子的扩散是各向异性的(anisotropic),水和液体和灰质是各向同性的(isotropic)。这很好理解,简单说就是纤维束中的分子运动只能朝着纤维束的方向扩散(轴突),脑脊液中的分子运动可以朝着周围随意运动,向各个方向运动的概率是相同的,这就是各项异性和各项同性的简单理解。这个是DTI分析的基本知识和背景。
所以其实纤维束构建的本质就是通过数学方法来计算Diffusion。说到计算,核磁里有一个技术,Pulsed-Gradient Spin-Echo Sequence(脉冲梯度自旋回波序列),就是在时间t内施加一个x方向的强磁场梯度,观察分子在前后运动的距离,就可以得出来弥散速度。

其实在临床上,我们看病人的核磁图象时,也能看到患者非弥散加权像-弥散加权像-两者比值这三张图,图的意义就是来自于上述思想。
因此,弥散速度的对比,我们可以通过两个变量来建模估算,一个是场强大小和场强方向。
场强大小,其实就是我们研究DTI像经常会看到的变量,即B值,B0,B1000,B3000等。

另一个就是方向:
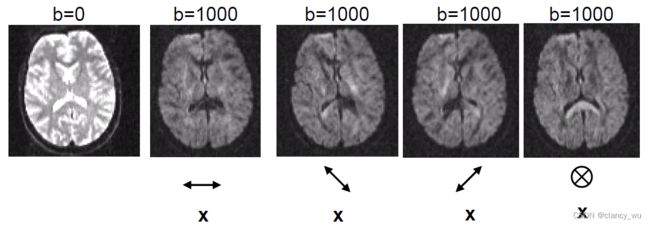
总结:
1.设置场强大小和方向,来估算Diffusion
2.白质是各项异性
3.脑脊液和灰质是各项同性
即,我们通过施加预设的场强大小和场强方向,来估算diffusion,算出来如果diffusion倾向于各项异性,我们就认为它可能是白质,反之则是灰质和脑脊液。
弥散张量成像 Diffusion Tensor Imaging,DTI
DTI的原理基础是弥散张量模型,Diffusion Tensor Model。即在每一个体素里计算它的 S j S_j Sj

在计算的时候,是按照定义的坐标系进行计算,XYZ坐标系:

其实还需要计算它的解剖学坐标系:
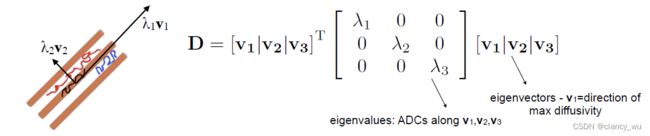
这样,通过这些定义的坐标系,我们就能用公式来表达各向异性和各向同性:

由此,可以得出各向异性的表达公式。各向异性的全称其实是Fractional Anisotropy,FA值,在[0, 1]之间。MD是平均扩散率

如下图:

不同场景对FA,MD的影响

细胞肿胀: F A ↑ , M D ↓ FA\uparrow,MD\downarrow FA↑,MD↓
髓鞘丢失: F A ↓ , M D ↑ FA\downarrow,MD\uparrow FA↓,MD↑
细胞死亡: F A ↓ , M D ↑ FA\downarrow,MD\uparrow FA↓,MD↑
这个也很好理解,结合上述原理就能理解。值得注意的是,FSL特别强调,这只是可能的几种解释情况,但是实际情况很复杂,所以不要过度解读。
但是,这里其实有一个缺点问题,暂时没办法解决,就是交叉纤维束(crossing fibers)和接触纤维束(kissing fibers)。不交叉的纤维束很好理解,它沿着轴突,计算出来的FA也很符合假设。但是在交叉的纤维束中,这个模型,它计算出来的FA反而是降低的。
另外,由于是单个体素的计算,每个体素都有一个值,因此需要有一个前提假设,就是一条纤维束的最大扩散率取值是主要纤维束上大部分体素的主要方向。这样不仅简化了计算,还使得值可以进行对比。
下面是常见的纤维束方向估计图:

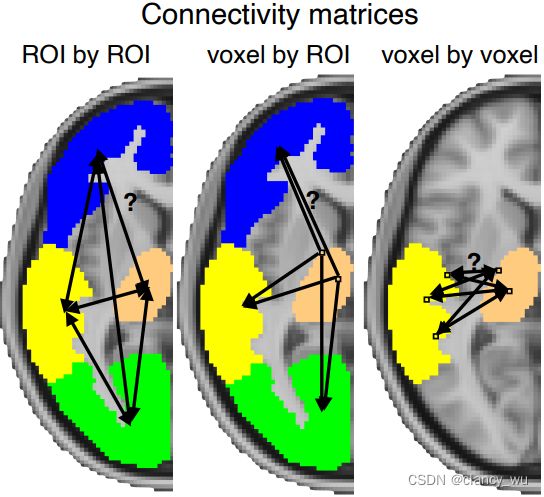
基于上述原理,衍生出几种常见的分析方法:
- Tract-Based Spatial Statistics TBSS
- VBM-style Analysis of FA
- voxel/ROI-based FA Comparison
- Tractography-Based FA Comparison
TBSS就是先预定义一个白质模板(比如霍普金斯白质张量模板),内有固定的白质纤维束,然后把原始数据在模板纤维束上进行计算FA,进行统计分析。
VBM就是在体素级别进行FA统计分析。
ROI就是在感兴趣脑区取主要体素的主要方向进行FA统计分析。
Tractography-Based就是在感兴趣ROI之间首先进行纤维成像,然后FA统计分析。
当然,原理虽然很清晰,但是实际应用中,还是会有一些问题,比如图象的失真/丢失,被试的头动等,都会对结果产生一定影响。这些就不是我们医学生关注的内容了。一般都会有固定的流程来尽量减少这些,例如eddy, topup等方法,还有mrtirx3的denoising, unringing等。
我们医学生一般关注的是有哪些现成的软件和已经经过验证的方法可以使用,从而发表自己的研究结果。
(以上内容,更具体的细节请参阅FSL官网资料)
MRtrix3
前面说到,有很多纤维束计算方法,TBSS是在固定模板上计算,又别称为确定性纤维成像,在FSL的BEDPOSTX模块可以完成;另外还有概率性纤维成像,就是追踪计算来构建纤维束,这个过程可以在FSL的PROBTRACKX模块可以完成。
除了FSL,其实还有另一种开源软件,MRtrix3,它在纤维追踪方面更为有优势和精确,dmriprep就是调用了FSL和MRtrix3模块来计算的。
前面提到,DTI的计算是基于弥散张量模型,Diffusion Tensor Model。这个模型在椭圆型张量表现很好,但是对于球形张量表现就很差,会出现FA下降的情况。球形张量包括比如crossing fibers和kissing fibers。因此,constrained spherical deconvolution CSD(约束球面反卷积)方法被提出用于解决crossing fibers的FA计算。而CSD方法就包括在MRtrix3里面。
基于CSD思想,MRtrix3提出了几种改善纤维束追踪的方法:
- Anatomically Constrained Tractography (ACT) — 解剖约束纤维束标记术
- Spherical-deconvolution informed filtering of tractograms (SIFT) — 纤维束的球面反卷积滤波
- Multi-shell Multi-tissue CSD (MSMT) — 多壳多组织的约束球面反卷积
ACT是根据解剖学基础,自动停止超出解剖学以外的不合理纤维束追踪。
SIFT是对纤维追踪中对于长纤维束容易出现高估的现象进行校正。
MSMT是根据不同组织对b值敏感性不同的现象来改善纤维束追踪,我的理解就是升维,在低维度下两者没有看出差别,那就提升维度,这样就会产生差别,有点类似SVM的思想。
MRtrix3教程
以下教程为初步使用,资料来源于Marlene Tahedl撰写的BATMAN
首先是准备数据,包括b1000, b2000, b3000的DWI图像和T1图像。
(一)预处理
1.合并图像,形成dwi数据
mrcat b1000_AP/ b2000_AP/ b3000_AP/ dwi_raw.mif
2.降噪
dwidenoise dwi_raw.mif dwi_den.mif -noise noise.mif
3.去除Gibb’s ringing伪影
mrdegibbs dwi_den.mif dwi_den_unr.mif -axis 0,1
axis 0,1 是说原始数据为axial slices, 0,2指coronal slices, 1,2指sagittal slices。
4.头动校正和失真校正 motion and distortion correction
dwiextract dwi_den_unr.mif - -bzero | mrmath - mean mean_b0_AP.mif -axis 3
mrconvert b0_PA/ - | mrmath - mean mean_b0_PA.mif -axis 3
mrcat mean_b0_AP.mif mean_b0_PA.mif -axis 3 b0_pair.mif
dwipreproc dwi_den_unr.mif dwi_den_unr_preproc.mif -pe_dir AP -rpe_pair -se_epi b0_pair.mif -eddy_options " --slm=linear"
这里一共有四步,解释一下,一个理解的难点在于distortion correction 失真校正(权威翻译我不指导叫什么,这个是百度翻译的结果),这个其实是针对EPI像的,由于人类鼻腔空气的影响,在扫描核磁的时候,鼻腔上块到额叶前端的部分经常会模糊,甚至丢失(非人为操作导致的,即使扫描的时候把额叶全部框进去了,还是会有这种误差,属于系统误差),这个时候就可以用distortion correction来尽量插添信号。而这个过程是通过两个相反方向磁场来实现的,就是b0_AP和b0_PA。所以第一步是在dwi里提取平均b0图象,因为dwi_den_unr的原始图象是AP方向扫描获取的,所以这个mean_b0就是AP方向。随后再计算b0_PA的平均值mean_b0_PA图象,然后用这两个AP-PA来合成b0_pair图象,用这个图象就能完成distortion correction。为什么用mean b0呢,作者认为mean值更干净,能减少误差,其实单纯用b0的AP和PA应该也行把(这个是我猜测的,可能会影响部分结果)。而motion correction就很简单,直接调用FSL的eddy函数。
值得一提的是,在FSL的标准处理中,是没有distortion这一步的,印象中只有MRtirx3有,所以好多文献是不做distortion这一步的。如果不做有什么影响呢?其实只会影响额叶前端一小块的bold signal,不会影响其他地方的纤维束追踪。
5.偏置场校正 Bias field correction
dwibiascorrect ants dwi_den)unr_preproc.mif
dwi_den_unr_preproc_unbiased.mif -bias bias.mif
这一步其实不是必须的。bias field correction的作用是改善brain mask的估算质量,但是呢,如果没有强偏置场而运行了这一步,则会降低brain mask的估算质量,所以这一步其实不是必须的。可以做可以不做。Marlene Tahedl在提供的练习数据中,小脑有一个明显的异常高信号,经过bias field correction后,小脑异常信号就降低了。但是,一般还是不太做,FSL反正是不做,我倒也不推荐做。
6.brain mask estimation
dwi2mask dwi_den_unr_preproc_unbiased.mif mask_den_unr_preproc_unb.mif
这一步没啥好说的,就是简单的brain mask生成。在FSL中会让你填threshold,这里不需要填,会根据默认值进行估算。
(二)Fiber orientation distribution 纤维方向分布 FOD
估算响应函数 response function estimation
经过上述的预处理后,就能开始进行纤维束相关的操作了。要进行这些操作,首先需要估算纤维束在每一个体素上的方向。前面说过,方向估算的常用模型是弥散张量模型,FSL中就是默认这个模型。而MRtrix3则是CSD方法(约束球面反卷积),CSD公式原理医学生不需要知道,CSD方法主要是通过Response Function(响应函数,RF)来完成反卷积过程,完成纤维束的方向估算。但是CSD方法还是有缺陷,就是CSD对单纯的白质部分效果不错,对于混杂部分效果不行,比如有的体素周围包括了脑脊液,灰质和白质等,不纯。这个时候可以用MSMT的方法,通过增加数个RF,来多个维度上对体素进行估算,前面说过,就类似于SVM(支持向量机)的升维过程,利用的是不同组织对不同场强敏感性不同的特性。
要完成MSMT过程,其实很简单,只要有multi-b值就可以,这个样例数据中有b1000, b2000, b3000,所以可以轻松实现。
dwi2response dhollander dwi_den_unr_preproc_unbiased.mif wm.txt gm.txt cfs.txt -voxels voxels.mif
生成的wm.txt, gm.txt, cfs.txt就是response function参数,为后面做准备。
纤维束方向计算Fiber orientation distribution estimation
dwi2fod msmt_csd dwi_den_unr_preproc_unbiased.mif -mask mask_den_unr_preproc_unb.mif wm.txt wmfod.mif gm.txt gmfod.mif cfs.txt cfsfod.mif
这一步很简单,所有的数据和必要参数准备完毕后,直接运行dwi2fod命令就好了。而纤维束估算的过程,就是根据RF来计算每个体素的方向,最后行成纤维束追踪。所以输入了wm.txt就输出了wmfod.mif,gm和cfs同理。
(三)Intensity Normalization 强度归一化
这一步主要是为了校正被试间差异,不同被试存在着fod生成方向的FA值的差异,比如可能由扫描仪器导致的,可能由其他原因导致的系统误差,因为在对所有被试进行一个总体的global normalization,从而减少系统误差,增强可比性。
mtnormalise wmfod.mif wmfod_norm.mif gmfod.mif gmfod_norm.mif cfsfod.mif cfsfod_norm.mif -mask mask_den_unr_preproc_unb.mif
(四) 创建全脑纤维束图
1.准备ACT
ACT的思想前面已经介绍了,ACT就是根据解剖学定位,去掉不合理生成的纤维束,比如脑脊液里肯定不会有纤维束。那么如何实现呢,就是用T1像,我们知道,T1像是最清晰的解剖图,所以可以把FOD生成的图对齐到T1像,然后进行修剪。
ACT过程需要两步,第一步是准备T1图象。这个T1跟我们核磁扫描得到的原始T1有点不同,这个T1要求是4个维度且标记有5种组织类型(皮层灰质,皮层下灰质,白质,脑脊液,病理组织—病理组织一般情况下是空值,例如中风后的病灶)。
mrconvert T1/ T1_raw.mif
5ttgen fsl T1_raw.mif 5tt_nocoreg.mif
就得到了解剖图,即5tt_nocoreg.mif
第二步是把解剖图对齐到dwi上,注意,对齐方向不能相反,因为dwi-T1会有很多信息损失,而T1-dwi则损失小很多。
dwiextract dwi_dem_unr_preproc_unbiased.mif - -bzero | mrmath - mean mean_b0_preprocessed,mif -axis 3
mrconvert mean_b0_preprocessed.mif mean_b0_preprocessed.nii.gz
mrconvert T1_raw.mif T1_raw.nii.gz
flirt -in mean_b0_preprocessed.nii.gz -ref T1_raw.nii.gz -dof 6 -omat diff2struct_fsl.mat
transformconvert diff2struct_fsl.mat mean_b0_preprocessed.nii.gz T1_raw.mif flirt_import diff2struct_mrtrix.txt
mrtransform T1_raw.mif -linear diff2struct_mrtrix.txt -inverse T1_coreg.mif
mrtransform 5tt_nocoreg.mif -linear diff2struct_mrtrix.txt -inverse 5tt_coreg.mif
虽然上述过程看起来很复杂,其实理解起来也简单。首先就是计算了mean_b0_preprocessed.mif,然后根据这个,使用FSL的FLIRT模块,生成了dwi到T1的转换公式矩阵,根据这个公式矩阵,就把第一步生成的有标记的T1像对齐到了dwi图象上。
2.生成mask
现在已经把T1像变换到了dwi图象上,所以就可以根据这个T1像来对生成的FOD纤维束图进行剪枝,去掉不合理的纤维束。
5tt2gmwmi 5tt_coreg.mif gmwmSeed_coreg.mif
3.生成纤维束图
tckgen -act 5tt_coreg.mif -backtrack -seed_gmwmi gmwmSeed_coreg.mif -select 10000000 wmfod_norm.mif tracks_10mio.tck
这个数字10000000指的是通过概率纤维成像,选择生成了这么多个纤维束,当然,其实也不需要这么多,可以用下面的命令减少一点,但是其实多点少点其实没太大差别
tckedit tracks_10mio.tck -number 200k smallerTracks_200k.tck
这样就只有200000个纤维束了。
4.进一步减少纤维束数量
前面在SIFT部分提到,很长的纤维束估算,其实是overestimated,超出了实际现象。这个是CSD算法的缺陷,因此Smith提出用SIFT算法来解决这个缺陷,减少过长纤维束的过度估算陷阱。当然,SIFT也有一个问题没办法解决,就是多长的纤维束才会开始overestimated。
tcksift -act 5tt_coreg.mif -term_number 1000000 tracks_10mio.tck wmfod_norm.mif sift_1mio.tck
在这里,作者是使用SIFT降低了10倍,这个其实是作者经验性定的,不是说10就是最佳的。事实上,在运行的时候,MRtrix可能会提示"quantisation error"的warning,这个warning出现代表MRtrix认为你减少的太多了,可能并建议重新定义下减少的数字。
ROI of tractograms
上面是全脑纤维束连接。假如我们只关注某个纤维束,例如皮质脊髓束,就需要用tckedit的include选项。
你可以通过ROI的mask来定义ROI,或者使用坐标(XYZ)来定义ROI,这样MRtrix就只会计算通过这个ROI的纤维束数量。
例如:
tcKedit -include -0.6,-16.5,-16.0,3 sift_1mio.tck cst.tck
当然,有一点需要强调,就是这个坐标(或者ROI mask),是真实坐标呢,还是模板坐标呢?要注意,与功能像不同的是,我们在上面所有处理中,都没有realign步骤,所以这里的坐标是真实坐标,而处理后的DWI图象和track 图象由于是T1对齐到DWI的,所以他们俩是同一个坐标系,用哪个图象去找坐标都可以。
还有一点提示,为了更好可视化,可以把纤维束图叠加到T1像上,还是使用diff2struct_mrtirx.txt这个转换参数,把T1_raw转换过去。可视化效果会好很多。
(五)structural-connectivity 结构连接
上面提了ROI的定量纤维束分析,其实还有种结构连接的分析,就是根据模板预定义的ROI,分析他们之间的纤维束连接。
1.选择模板
这里作者选择的是人类连接组项目中,Glasser发表的HCP MMP 模板。需要在Freesurfer上对这个模板进行脑区分割。
mrconvert T1_raw.mif T1_raw.nii.gz
recon-all -s snake -i T1_raw.nii.gz -all
mri_surf2surf --srcsubject fsaverage --trgsubject snake --hemi lh --sval-annot fsaverage/label/lh.glasser.annot --tval snake/label/lh.hcpmmp1.annot
mri_surf2surf --srcsubject fsaverage --trgsubject snake --hemi rh --sval-annot fsaverage/label/lh.glasser.annot --tval snake/label/lh.hcpmmp1.annot
#snake是作者自定义的名字
2.生成矩阵
tck2connectome -symmetric -zero_diagonal -scale_invnodevol sift_1mio.tck hcpmmp1_parcels_coreg.mif hcpmmp1.csv -out_assignment assignments_hcpmmp1.csv
这个csv文件就是所有脑区的脑区间连接矩阵。
3.ROI
ROI分析有两种,ROI-ROI和ROI-seed,即一种是两个ROI之间的structural connectivity,另一种是单个ROI中,所有穿过的纤维束。
第一种:
connectome2tck -nodes 8,188 -exclusive sift_1mio.tck assignments_hcpmmp1.csv moto
图象合并成一个文件
mrcalc roi_1.mif roi_2.mif -max merged_roi.mif
第二种:
connectome2tck -nodes 362,372 sift_1mio.tck assignments_hcpmmp1.csv -files per_node thalamus
注意,-files per_nodes一定要选,不然所有纤维束会以单个文件生成,形成成千上万个文件,所以需要合并成一个文件,就是per_node。
一些补充知识点:
node换成mesh,可视化:
label2mesh hcpmmp1_parcels_coreg.mif hcpmmp1_mesh.obj
这样脑区就不是节点的形式呈现,而是脑区的形状呈现。
连接呈现:
connectome2tck sift_1mio.tck assignments_hcpmmp1.csv exemplar -files single -exemplars hcpmmp1_parcels_coreg.mif
注意,这里的exemplar是自定义的,是输出文件的前缀名称,例如这里生成的是exemplar.tck文件。这可以显示出两个ROI的连接路径,是怎样的形式来连接的。
