微生物共现网络可视化:实现布局自由
ggraph包常用于网络数据可视化,其语法特征与ggplot2相同,我们很容易将ggplot2的知识扩展到ggraph上。ggraph的绘图思路大致如下:
- 计算网络(
graph)的布局,获得节点(vertex)在X-Y轴上的坐标值,返回布局数据框layout_df; - 利用1中返回的数据框
layout_df,利用ggraph(layout_df)函数绘制画布; - 添加图形元素,即节点(
vertex)和边(edge)。
在计算网络布局中,可以使用已经有的算法计算布局,例如"fr", "cycle", "stress"等,但有时我们希望能根据自身数据的需要进行布局,那么我们就可以使用 layout_df <- create_layout(graph_test,layout="manual",x=x,y=y)计算出网络文件graph_test的布局数据框layout_df,然后像在ggplot2中绘图一样绘制网络图即可。
layout_df <- create_layout(graph_test,layout="manual",x=x,y=y)
ggraph(layout_df)+
geom_edge_link()+
geom_node_point()
重点就是:如何根据自身需要计算出节点(vertex)在X-Y轴上的坐标值x和y。
譬如,我利用stress布局获得了如下结果:

但我还是觉得有些混乱,我想将同一门内的微生物放在一块进行可视化,如下图:
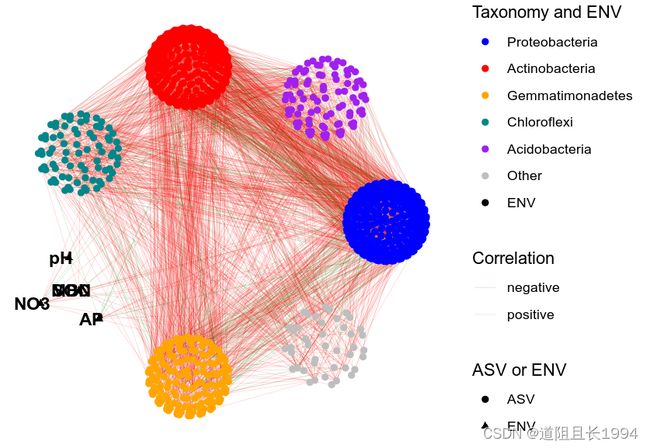
此时,我们可以根据自身的需要,巧妙地计算各个节点对应的X-Y值,然后自定义布局即可。虽然我们是自定义布局,但我们也没有必要从轮子造起,灵活运用已有的布局算法实现我们的想法,思路如下:
- 根据节点的分类信息属性将网络分解为若干个子网络,即
Proteobactria network,Acidobacteria network等; - 对各个子网络利用
sphere算法计算网络布局 - 对获得的布局坐标进行圆形偏移,即获得自定义布局的XY坐标
- 根据
xy值自定义网络布局
实战代码
下面我们以按模块分类为例,给出其大致的思路及具体的R代码。
- 总体思路:
- 对微生物共现网络的模块进行划分;
- 按照模块大小排序,对前面的模块进行着色,其余归为其它;
- 对各模块进行圆形布局,求解布局坐标.
- R代码:
library(magrittr)
library(igraph)
library(ggraph)
library(purrr)
g1 <- read_graph("~/Desktop/graph_test.graphml",format = "graphml")
wtc <- cluster_louvain(g1,NA)
modularity(wtc)
m<-membership(wtc)
sorted_table_m <- sort(table(m),decreasing = TRUE)
top <- 1:5
df1 <- data.frame(old=names(sorted_table_m),new=c(top,rep("other",length(sorted_table_m)-length(top))))
new_m <- unlist(map(m,function(x) df1[df1$old%in%x,2]))
groupVector <- function(x){
lev <- levels(as.factor(x))
len <- length(lev)
list1 <- vector("list", len)
for(i in 1:len){
list1[[i]] <- names(x)[x == lev[i]]
names(list1)[i] <- lev[i]
}
return(list1)
}
gv <- groupVector(new_m)
g_list <- map(gv,~induced_subgraph(g1,.x))
ly3 <- map(g_list,function(x) create_layout(x,layout = "sphere"))
x <- ly3 %>% map(~.$x)
y <- ly3 %>% map(~.$y)
theta <- seq(0,2*pi,2*pi/(length(g_list)))
mx <- 4*cos(theta[-1])
my <- 4*sin(theta[-1])
mxx <- unlist(map2(x,mx,function(x,y) x+y))
myy <- unlist(map2(y,my,function(x,y) x+y))
name <- unlist(ly3 %>% map(~.$name))
df2 <- data.frame(x=mxx,y=myy,row.names = name)
ly4 <- create_layout(g1,layout = "manual",x=df2[V(g1)$name,1],y=df2[V(g1)$name,2])
edgeColor <- ifelse(E(g1)$weight_1>0,"positive","negative")
colorfactor <- factor(new_m[ly4$name])
mycolor <- c("blue","red","orange","turquoise4","purple","gray")
ggv4 <- ggraph(ly4)+
geom_edge_link(aes(colour=as.factor(edgeColor)),edge_width=0.1,alpha=0.3)+
geom_node_point(aes(color=colorfactor))+
theme_minimal()+
scale_edge_color_manual(values = c("green4","red"),name="Correlation")+
scale_color_manual(values = mycolor,name="Modular")+
theme(axis.title = element_blank(),
axis.ticks = element_blank(),
axis.line = element_blank(),
axis.text = element_blank(),
panel.grid = element_blank())
ggv4
pdf("~/Desktop/graph visualization by modular.pdf",width = 10,height = 7)
ggv4
dev.off()
- 实现效果
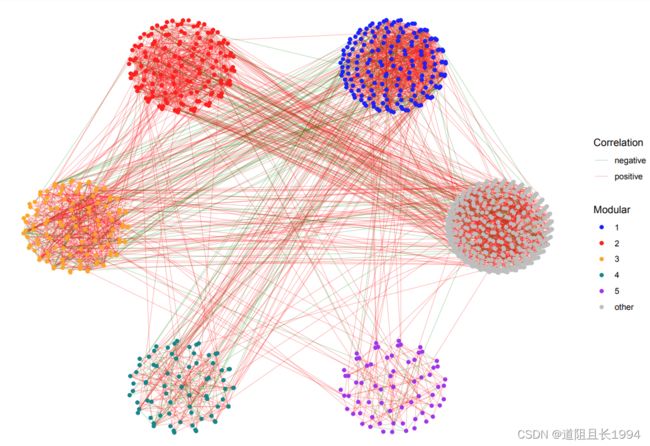
测试数据可查看:
- 微生物共现网络可视化:按模块进行圆形布局
- 微生物与环境因子共现网络可视化:按物种分类着色,并进行圆形布局