MATHLA:集成双向LSTM和多头部注意力机制的HLA-肽结合预测的强大框架
我们提出了一种整合了双向长短期记忆网络和多头注意力机制的泛等位基因HLA-肽结合预测框架——MATHLA。该模型在五倍交叉验证测试和独立测试数据集中都实现了更好的预测准确性。此外,该模型在预测11至15个氨基酸的较长配体的准确性方面优于现有工具。此外,我们的模型还显示了HLA-C-肽结合预测的显着改进。通过研究多头注意力权重评分,我们描述了三个HLA I超群与其同源肽之间可能的相互作用模式。
最近,生成了95个HLA I类等位基因的更大质谱数据,这进一步说明了通过将更多数据纳入模型来提高预测准确性[7]。然而,数据依赖性方法受到当前技术可以处理的等位基因数量有限以及三个HLA I类超群(A,B和C)之间数据条目不平衡的限制。因此,由于鉴定的HLA-C配体数量较少,数据依赖性方法往往导致HLA-C等位基因的预测准确性降低。
泛等位基因模型的原理是,可以明确勾勒HLA等位基因的核心序列[8]。因此,作为HLA配体序列,核心HLA序列可以被编码并馈送到学习算法中,用于模拟HLA-肽相互作用。此外,由于HLA的大多数天然配体长度为8-11个氨基酸[11],因此训练数据集中其他长度的配体数量有限将影响预测HLA结合的12mer至15mer的准确性[12]。因此,非常需要建立一个更稳健的泛等位基因模型,该模型能够以更高的精度预测更长长度的配体和HLA-C等位基因的同源肽。
本文提出了一种新颖的深度学习HLA-表位结合预测方法,该方法利用双向LSTM从较长序列中提取信息的内在能力和多头注意力机制从不同角度捕获上下文依赖性的能力。所提出的框架在预测HLA-C等位基因和肽之间的结合方面更强大。此外,该框架在预测长度为12至15个氨基酸的配体方面比现有工具更强大。最后,该模型还可以通过相互作用表示的多个亚空间帮助解释HLA等位基因和肽之间的相互作用。
数据
为了将定性亲和力数据转化为定量值,我们应用了与MHCflurry [3]相似的规则:阳性-高,< 100nM;阳性,< 500纳米,阳性-中间,< 1000纳米;正-低,< 5000nM;负值,> 5000纳米;质谱鉴定的配体,< 500纳米;诱饵,> 5000海里。此外,我们应用了另一套规则来消除相同等位基因-肽对的测量冗余:如果其他数据是通过不等式测量的,则保留带有“=”的唯一数据;如果所有数据都用“>”测量,则保留具有最大亲和力值的数据;如果所有数据都用“<”测量,则保留亲和力值最低的数据;并且丢弃具有矛盾测量的所有剩余数据。
为了便于模型训练,我们将原始纳摩尔亲和力标准化为0到1。
![]()
其中anormal是归一化亲和力,anM是原始纳摩尔亲和力值。最终的训练数据集由167个HLA I类等位基因(53个HLA a、92个HLA-Bs和22个HLA-Cs)的753,961个条目组成。
测试数据集
测试数据集的阳性数据来自最近的大规模HLA I类配体组数据,包括95个HLA等位基因。来自16个HLA等位基因的数据条目先前由同一组产生[16]并包括在训练数据集中,首先被排除。接下来,我们保留了长度为8至15个氨基酸的HLA展示配体,并去除了那些具有翻译后修饰的配体。为了将阴性数据引入到测试数据集中,我们随机取样诱饵肽序列,这些序列不包括在来自阳性肽的宿主蛋白编码转录物的阳性数据集中。对于每个阳性肽,相应地产生100个诱饵序列。最后,在过滤掉与训练数据集重叠的数据条目之后,在测试数据集中总共有140,232个阳性肽和13,939,114个阴性诱饵。
网络模型
根据BLOSUM62置换矩阵[17],将肽和HLA伪序列的每个残基(从netMHCpan 4.0 [9]检索)编码为相似性得分向量。(残基:在蛋白质的序列中,氨基酸之间的氨基和羧基脱水成键,而剩下的没有脱水成键的基团就叫残基。)
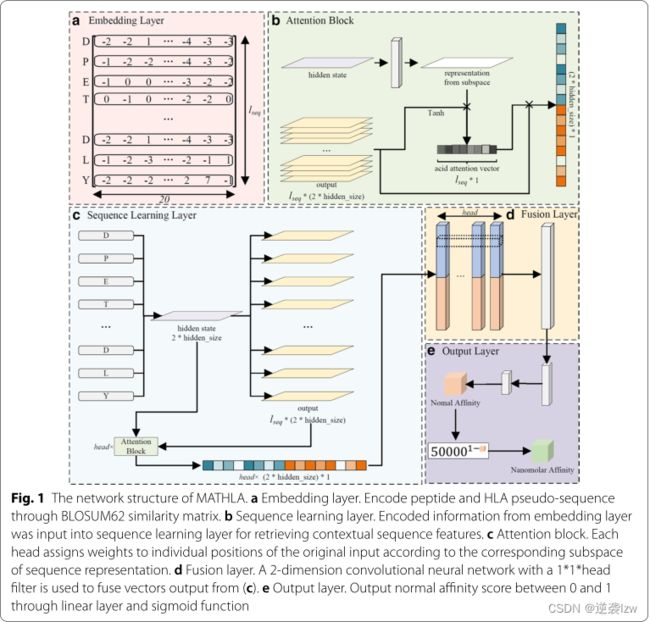
(图中标注有误,正确顺序:a--c--b--d--e)
图1 math la的网络结构。a嵌入层。通过BLOSUM62相似性矩阵编码肽和HLA伪序列。c序列学习层。来自嵌入层的编码信息被输入到序列学习层,用于检索上下文序列特征。b注意块。每个头根据序列表示的相应子空间将权重分配给原始输入的各个位置。d融合层。具有1 * 1 *头部滤波器的二维卷积神经网络用于融合从(c)输出的向量。e输出层。通过线性层和sigmoid函数输出介于0和1之间的正常亲和力得分
(图1 a)与许多其他方法不同,我们的模型使用预定义的“填充”规则来确保训练过程中输入矩阵的等维,允许输入序列具有灵活的长度。维数为lseq*20的编码矩阵,其中lseq是级联的长度然后将肽序列和HLA伪序列输入序列学习层
(图1 c)我们选择了BILSTM[18]来模拟具有灵活长度的多肽的氨基酸残基之间的依赖关系。与传统的递归神经网络相比,带有门控制单元(输入门、遗忘门和输出门)的LSTM网络能够更有效地学习肽序列中距离较远的残基之间的依赖信息。为了增强我们的模型学习n端和c端氨基酸残基之间的双向依赖的能力,使用了双向LSTM(bi-LSTM)[19]。通过将正向序列和反向序列分别输入到具有相同结构的LSTM网络,得到了正向序列和反向序列在时间t的LSTM ht和h‘t的输出,并将t位的人类白细胞抗原多肽序列表示为![]() 最后,将bi-LSTM的输出表示为outLstm。
最后,将bi-LSTM的输出表示为outLstm。
LSTM传送门:长短时记忆网络 LSTM — PaddleEdu documentation
BILSTM传送门:Bi-LSTM学习_一枚小白的日常的博客-CSDN博客_bi-lstm
(图1 b) 此部分为c中注意力块,上面部分第一个输入为要进行查询的点的向量表示(为注意力机制中的Q),之后为和从多头注意力中选取其中一个,下面输入的output为b中得到的各个氨基酸的向量表示,为注意力机制中的K。为了处理来自序列表示不同子空间的不同位置的肽信息,我们将多头注意机制[20,21]应用于双向LSTM的输出。
多头注意力机制传送门:This post is all you need(①多头注意力机制原理) – 月来客栈


其中![]() 是双LSTM网络的隐状态
是双LSTM网络的隐状态
![]() 是将原始的隐状态投影到不同的表示子空间的权重
是将原始的隐状态投影到不同的表示子空间的权重
![]() 是注意权重
是注意权重
(·)T表示矩阵的转置
![]() 是上下文向量
是上下文向量
Outlstm∈rlseq×(隐藏×2)是LSTM网络的输出
![]() 是第i个注意机制下原始序列的注意向量
是第i个注意机制下原始序列的注意向量
合并前向和后向注意的级联输出向量。然后,通过学习每个注意力头部的权重,将具有头部*1*1过滤器(头部*1*1滤波器后的效果就是将各个向量对应位置处的值相加,放入最后向量对应位置)的二维卷积神经网络(2D CNN)应用于融合向量的组合向量。
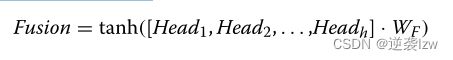
其中h表示多头注意模块的头数,WF∈Rh×1×1是2D CNN的滤波器,Fusion∈r1×(Hidden×2)是应用2D CNN后的输出向量。最后,通过应用具有S型激活函数的线性层(图1E),输出从0到1的预测值。

其中Wo∈R(隐藏×2)×1和b分别是线性层的权重向量和偏差。
为了最大限度地减少异常值(噪声)对模型训练的影响,我们在训练过程中采用了优化的Huber损失函数[22]。
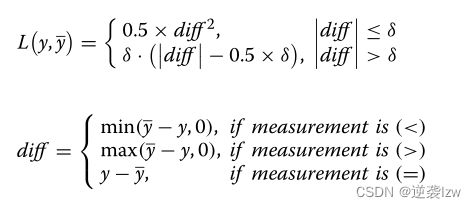
其中y和y分别是观察和预测的结合亲和力的值。当y和y之间的不平等关系不满足时,它们的差异(Diff)将因此影响损失。当差值小于期望值δ时,胡伯损失退化为均方误差损失。另外,Huber Lost使用线性误差来评估训练损失,从而能够最大限度地减少难以学习的数据对模型训练性能的影响。采用Radam方法对模型参数进行了优化。与传统的ADAM相比,Radam[23]能够调整自适应学习速率的方差,从而防止模型收敛到局部极小值。批大小被选择为512,并且训练停止,因为验证损失数据集在连续5个时期之后没有改善。纪元数设置为100。学习速率被设置为0.001,并且辍学率被设置为0.1.
结果:
与其他模型相比,MATHL A的改善程度与多肽的长度呈正相关(图2)。2B)。特别是对于长度为12~15个氨基酸的较长多肽,MATHLAAUC平均得分为0.926
MATHLA在新等位基因上的表现优于现有的泛等位基因模型
为了测试MATHLA的泛化能力,我们利用了我们的训练数据集和一组质谱学的HLA配子组数据之间不重叠的等位基因的优势总共,95个等位基因中有10个用于评估泛等位基因模型的泛化能力。另外两个泛等位基因模型-netMHCpan4.0和ACME用于模型比较,无论这10个等位基因是否包括在他们的训练数据集中(ACME只支持7个HLA-A和HLA-B等位基因)。总体而言,MATHLA在非重叠等位基因中的表现分别比netMHCpan4.0和ACME高80%和100%。我们的模型超过10个等位基因的平均AUC值达到0.982,高于netMHCpan4.0(图3A)的0.975。与netMHCpan4.0相比,值得注意的是,相比于HLA-A和HLA-B等位基因,MATHLA的性能优势在HLA-C等位基因上更为突出。三个人类白细胞抗原C等位基因的平均AUC值在MATHLA4组为0.988,而在netMHCpan4.0组为0.965。
MATHLA提高了现有模型对HLA-C等位基因的准确性
但与HLAC组相对应的MATHLAAUC值明显高于竞争模型(MATHLA4.0为0.976,netMHCpan4.0为0.951,MHCflries为0.927)(图3C)我们证明了在有限的训练数据的情况下,我们的模型可以比泛等位基因和特定等位基因的模型更好地识别人类白细胞抗原-C等位基因。
MATHLA能够描述各种人类白细胞抗原-配体结合模式
以前对HLA配体的基序分析表明,最C末端的残基比其他位置最有可能具有经常性氨基酸。
我们发现由9个氨基酸组成的多肽的重量分布与所有其他长度的多肽不同,其中第9位的重量占主导地位。这一模式与之前的发现一致,即C末端残基对9-肽的结合比更长的肽更重要
与人类白细胞抗原-A和B结合的多肽的第二或第三个位置的注意力权重分数与另一个已知基序一致,而人类白细胞抗原-C配体的相应位置所占的权重要小得多。这种独特的人类白细胞抗原C配基的重量模式可能解释了为什么我们的模型在预测人类白细胞抗原C肽方面比其他工具有更大的优势
总结
我们的模型结合了双向LSTM和多头注意机制,解决了这两个问题,不仅在预测HLA-C等位基因方面取得了显着的优势,而且对较长的I类HLA配体也获得了更好的预测能力。我们的工作表明,先进的深度学习体系结构可以为进一步改进和理解HLA-肽结合预测提供一个解释性模型。我们预计,引入其他替代方法,如自我注意机制和word2vec模型,可以提供更好的肽表示,以进一步提高预测精度。我们的框架肯定将有利于基于T细胞的疫苗的开发,用于治疗癌症和预防传染病。