论文解读:《一种基于长短期记忆网络深度学习的药物靶相互作用预测方法》
论文解读:《A deep learning-based method for drug-target interaction prediction based on long short-term memory neural network》
- 略缩词
- 1.文章概括
- 2.背景
- 2.材料和方法
-
- 2.1 数据收集
- 2.2 药物分子的表征
- 2.3 目标蛋白的表征位置特异性得分矩阵(PSSM)
- 2.4 勒让德矩阵(Legendre moments,LM)
- 2.5 特征压缩和融合
- 2.6 构造DeepLSTM模型
- 2.7 防止过拟合
- 3.实验设定
-
- 3.1 评价指标
- 3.2 模型训练
- 4.结果与讨论
-
- 4.1 与其他分类器模型的比较
- 4.2 与最先进的方法进行比较
- 5.讨论
文章地址:https://bmcmedinformdecismak.biomedcentral.com/articles/10.1186/s12911-020-1052-0
DOI:https://doi.org/10.1186/s12911-020-1052-0
数据:https://Deepbiolab.coding.net/s/fbdb894d-2730-425b-bac6-18ba55396bab
代码:https://deepbiolab.coding.net/s/fbdb894d-2730-425b-bac6-18ba55396bab
略缩词
ACC:准确度(Accuracy)
DeepLSTM:深度长短期记忆网络(Deep long short-term memory)
DTIs:药物-靶标相互作用(Drug-target interactions)
LMs:勒让德矩阵(Legendre moments)
MCC:马修斯相关系数(Matthew’s correlation coefficient)
MLP:多层感知器(Multi-layer perceptron)
PPV:阳性预测器(Positive predictive value)=TP/(TP+FP)*100%
PSSM:特定位置计分矩阵(Position specific scoring matrix)
RNN:循环神经网络(Recurrent neural network)
SPC:特异性(Specificity)
SPCA:稀疏主成分分析(Sparse principal component analysis)
SVM:支持向量机(Support vector machine)
TPR:真阳率(True positive rate)
1.文章概括
- 背景:现代药物发现的关键是发现,识别和准备药物分子靶标。但是,由于通量,精度和成本的影响,传统的实验方法很难广泛用于推断这些潜在的药物-靶标相互作用(DTI)。因此,迫切需要开发有效的计算方法来验证药物与靶标之间的相互作用。
- 方法:作者开发了基于深度学习的DTI预测模型。蛋白质的进化特征是通过特定位置评分矩阵(PSSM)和勒让德矩阵(LM)提取的,并与药物分子的亚结构指纹相关联,以形成药物-靶对的特征向量。然后,利用稀疏主成分分析(SPCA)将药物和蛋白质的特征压缩到统一的向量空间中。最后,构建了深长短期记忆(DeepLSTM)以进行预测。
- 结果:在实验结果上可以观察到DTI预测性能的显着改善,在四个重要的药物目标数据集上的AUC分别为0.9951、0.9705、0.9951和0.9206。进一步的实验初步证明,提出的特征描述方案在特征表达和识别方面具有很大的优势。作者还表明,所提出的方法可以很好地用于小型数据集。
- 结论:结果表明,与最新的药物靶标预测器相比,该方法具有很大的优势。据作者所知,此研究首先测试了具有记忆和图灵完整性的深度学习方法在DTI预测中的潜力。
2.背景
药物靶标是药物研究和开发的基础,在过去的几个世纪中,人们严重依赖目前已知用于检测药物的数百种药物靶标。尽管与靶蛋白相互作用的已知药物的数量继续增加,但是批准的药物靶的数量仍然只是人类蛋白质组的一小部分。检测药物与靶标之间的相互作用是开发新药物的第一步,也是药物筛选和药物定向合成的关键因素之一。得益于高通量实验,人们对药物化合物的结构空间和靶蛋白的基因组空间有了越来越多的了解。不幸的是,由于耗时费力的实验过程,我们对两个空间之间关系的理解仍然相当有限。由于公开提供的生物和化学数据的迅速增加,研究人员可以通过计算方法来系统地学习和分析异类新数据,并重新审视药物-靶标相互作用(DTI)。有几个专注于药物与靶标之间关系的免费数据库,例如ChEMBL,DrugBank,SuperTarget。这些数据库内容构成了黄金标准数据集,对于开发预测DTI的计算方法至关重要。
目前,用于DTI预测的计算方法可以分为三类:基于配体的方法,对接方法和特征学习方法。基于配体的方法通常用于通过计算给定药物或化合物与已知靶标活性化合物的化学结构相似性来估计潜在的作用靶标。 Keizer等人提出了一种基于蛋白质配体的化学相似性推断蛋白质靶标的方法。Yamanishi等人通过将化合物的化学结构相似性和蛋白质的氨基酸序列相似性整合到一个统一的空间中来预测未知的药物-靶标相互作用。 Campillos等人通过表型副作用的相似性预测潜在的靶蛋白。这种基于配体的方法在化学结构相似性高的情况下既简单又有效,但在很大程度上也限制了其应用范围和准确性。对接方法是计算药物和三维结构中潜在靶标的形状和电匹配,从而推断出药物作用的可能靶标。其中,反向对接方法是最常用的预测方法。该方法通过预测给定化合物与靶标之间的相互作用模式和亲和力来对药物靶标进行排名,从而确定药物的可能靶标。Cheng等人开发了一个基于结构的最大亲和力模型。 Li等人开发了一个名为TarFisDock的网络服务器,该服务器使用对接方法来识别毒品目标。这些方法充分考虑了目标蛋白质的三维结构信息,但是分子对接方法本身仍存在一些尚未得到有效解决的问题,例如蛋白质的柔韧性,评分功能的准确性以及溶剂水分子,导致反向对接。该方法的预测精度低。对接的另一个严重问题是它不能应用于具有未知3D结构的蛋白质。到目前为止,具有已知3D结构的蛋白质仍然只是所有蛋白质的一小部分。这严重限制了该方法的推广和普及。特征学习方法将药物靶标关系视为两类问题:相互作用和非相互作用。此类方法使用机器学习算法学习已知化合物目标对的潜在模式,通过迭代优化生成预测模型,然后推断潜在的DTI。 Yu等人提出了一种基于化学,基因组和药理学信息的系统方法。 Faulon等人使用签名分子描述符预测药物靶标。尽管这些方法加快了药物靶标的发现,但仍有很大的改进空间。
在这项工作中,我们提出了一种基于深度学习的方法来识别未知的DTI。所提出的方法包括三个步骤:(i)代表药物靶标对。药物分子被编码为指纹特征,而蛋白质序列特征则通过使用包含关于蛋白质进化信息的位置特异性得分矩阵(PSSM)上的Legendre Moments(LM)获得。(ii)特征压缩和融合。稀疏主成分分析(SPCA)用于减少特征维和信息冗余。(iii)预测。采用深长期短期记忆(DeepLSTM)模型来执行预测任务。作者提出的模型的流程如图1所示。在涉及酶,离子通道,GPCR和核受体的四个重要DTI数据集上实施了提出的方法。暴露的结果为现有的DTI预测最新算法提供了卓越的性能。
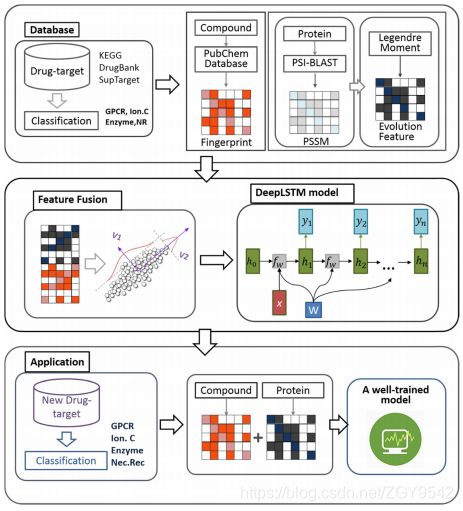
2.材料和方法
2.1 数据收集
作者通过KEGG,DrugBank 和SuperTarget数据库收集了有关药物化合物与靶蛋白之间相互作用的信息。表1根据药物化合物的数量,目标蛋白和相互作用总结了数据集。这套已知的DTI被认为是评估所提出方法性能的金标准。靶蛋白与药物分子连接形成药物靶网络。为了从网络获得阳性数据集,将金标准数据集中所有已识别的药物-靶标对都视为阳性样品。阴性样品对应于网络中其余的药物-靶标对。由于非交互对的规模远大于交互对的规模,因此构造的数据集不平衡。为了解决数据集不平衡引起的偏差,我们从网络中剩余的药物靶标对中随机选择阴性样品,直到阴性样品的数量与阳性样品的数量相同。

2.2 药物分子的表征
在一些研究中已经证实了亚结构指纹表征药物分子的能力。通过对先前研究结果的综合分析,PubChem指纹用于表征每种药物分子。在这项工作中,药物是编码的boolean亚结构向量,表示一个分子中是否存在相应的亚结构。 PubChem数据库定义了881个化学子结构,其中每个子结构都分配给特定的位置。因此,对于在药物化合物中出现的子结构,将指纹矢量中与该子结构相对应的位置设置为1,否则将相应位置设置为0。因此,每种药物均表示为881维矢量。
2.3 目标蛋白的表征位置特异性得分矩阵(PSSM)
位置特异性评分矩阵(PSSM)是查找远距离相关的蛋白质。近年来,PSSM被广泛用于蛋白质组学和基因组学研究,例如预测DNA或RNA结合位点和膜蛋白类型。在本文中,PSSM用于编码蛋白质并获得有关氨基酸的进化信息。具有N个氨基酸残基的蛋白A的PSSM可以表示为:
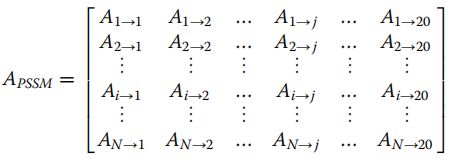
其中Ai→j是代表第i个残基突变为第j个天然氨基酸的概率的得分,N是序列A的氨基酸残基的长度,其中20表示20种天然氨基酸类型。每个蛋白质序列得到PSSM,都使用了位置特异性迭代BLAST(PSI-BLAST),并且选择了默认参数(除了三个迭代之外)。
2.4 勒让德矩阵(Legendre moments,LM)
不变矩是全局统计特征,并且在尺寸不变性,旋转不变性和位移不变性方面具有优异的特征,这有助于提取稳定性特征。勒让德矩阵(LM)是一种快速不变矩特征提取技术,在许多模式识别(即图形分析,目标识别,图像处理,分类和预测)的应用中显示出良好的性能。作者使用Legendre矩进一步细化PSSM中包含的进化信息并生成特征向量。 LM是连续的正交矩,可用于表示具有最小信息冗余的对象。顺序为(a,b)的LM定义为:
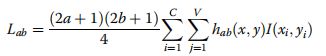
其中I(x,y)是一组离散点(xi,yi),xi,yi∈[-1,+1]。在本文中,I(x,y)表示PSSM,C是PSSM的行数,V表示PSSM每一列的总和。
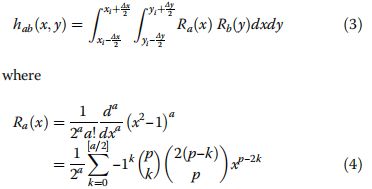
公式(3)中的积分项通常通过零阶近似来估计,勒让德多项式的值在[xi-Δx/2,xi+Δx/2]和[yi-Δx/2,yi+Δx/2 ]。因此,近似LM的集合定义为:
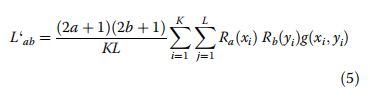
结果,在蛋白质序列的PSSM上使用LM,通过设置a,b = 30,从每个蛋白质序列中获得了961个特征。
2.5 特征压缩和融合
通过将药物亚结构指纹特征(881-D)与蛋白质LMs特征(961-D)相结合,作者从每个药物靶对中获得了1842维的药物靶特征向量。为了节省分类器的计算时间,减少内存消耗并消除原始特征空间中的嘈杂特征,稀疏主成分分析(SPCA)用于将药物和目标蛋白的特征整合到一个有机整体中,减小特征维数和冗余度信息。经典的主成分分析(PCA)有一个明显的缺点,即每个PC是所有变量的线性组合,并且载荷通常为非零。因此,当处理两种不同类型的特征(例如本文产生的药物和蛋白质特征)的组合时,通常会导致不可预测的结果。 SPCA是一种改进的PCA,它使用套索(弹性网)来生产具有稀疏载荷的主成分,从而克服了上述问题。最后,作者获得了400维精炼特征向量作为分类器的输入。
2.6 构造DeepLSTM模型
与标准RNN网络的主要区别之一是LSTM体系结构使用内存块代替求和单元。如图2所示,存储块包含用于存储时间状态的自连接存储单元,以及用于控制信息流的门(特殊乘法单元),输入门,输出门和遗忘门。这些门使LSTM能够在很长的一段时间内进行存储和访问,从而减少了梯度问题对预测模型的影响。进入存储单元的输入激活流程由输入门控制。单元激活的输出流流向网络的其他部分,该部分受输出门控制。通过单元的自递归连接,将遗忘门作为输入添加到单元中,以便LSTM网络可以处理连续的输入流。此外,LSTM单元可以包括窥孔连接,这些孔允许根据内部存储器中的状态值来调制门。
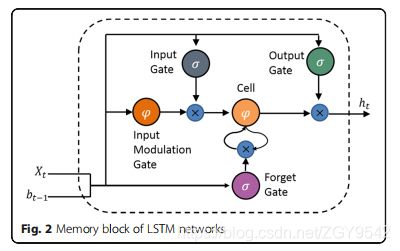
作者通过堆叠多个LSTM层来构建DeepLSTM,与简单的三层体系结构相比,深度体系结构可以通过在空间中多层分布更好地使用参数。深度导致输入在每个时间步长中经历更多的非线性运算。
2.7 防止过拟合
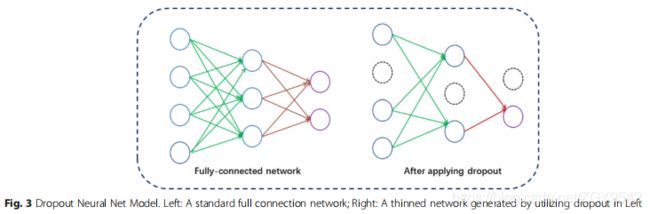
dropout用于解决过度拟合问题,方法是从神经网络中随机删除各个单元及其在训练过程中的连接。 “Dropout”的意思是从原始网络中提取一个“稀疏”网络,该稀疏网络由所有尚存的单元组成,如图3所示。在本文中,作者按照先前的研究来设置dropout率降至0.5。有35个隐藏层单元,在训练过程中可能会生成235个不同的子网。在测试阶段,采用了“平均网络”策略,该策略包含所有原始网络连接,但是它们的传出权重减半,以弥补其中活动的两倍多的事实。
3.实验设定
3.1 评价指标

3.2 模型训练
对于四个数据集,将每个数据集分为:训练集(80%)、验证集(10%)、测试集(10%)。使用训练集来拟合DeepLSTM预测模型,使用验证集来优化DeepLSTM神经网络权重,使用测试集以验证模型性能。使用验证集的另一个好处是可以通过提前停止来防止过度拟合:当验证数据集上的错误不再减少并且趋势增加时,终止模型训练。该技巧可避免过拟合并降低模型的训练成本。
对单元格输入单元和单元格输出单元使用双曲正切激活,对输入,输出和忘记门单元使用逻辑Sigmoid。 LSTM和RNN的输入是40维特征。输出层是一个完全连接的网络,并使用softmax函数生成概率结果。为了找到最佳的网络结构,我们在验证数据上测试了具有不同层数和单位的DeepLSTM模型的性能。从1到6进行试验的隐藏层数。关于单位数,以步长s = 4从20到200进行试验。最后,确定具有4个隐藏层和36个单位的DeepLSTM模型。 DeepLSTM的权重使用均值为0且标准差为0.1的随机数初始化。使用初始值0.002,衰减0.004和动量0.5的动态学习率,用均方误差和Nadam优化器训练模型。时间步长设置为1,批处理大小为64。最多进行500次迭代后停止训练,或者如果验证数据上没有新的最佳错误,则提前停止训练。
4.结果与讨论
表2给出了所提出模型的预测性能的统计数据。
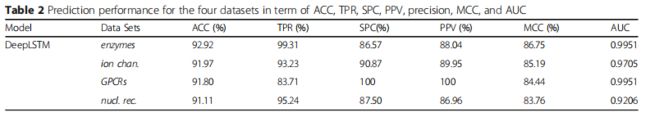
作者的方法仅使用180个样本就可以在核受体数据集上实现90%以上的准确性,在非常小的训练样本的情况下,作者的方法可以提供出色的性能。非凡的表现主要来自以下三个方面:(1)作者的特征表示方法可以有效地从药物分子和靶蛋白序列中提取出鉴别特征;(2)SPCA在计算效率,高解释方差和识别重要变量的能力等多个方面均具有优势,它将两个不同的特征向量压缩为统一的特征空间并提取异构特征;(3)分层结构使神经网络能够将输入数据转换为新的特征空间,从而更有利于完成分类任务。
4.1 与其他分类器模型的比较
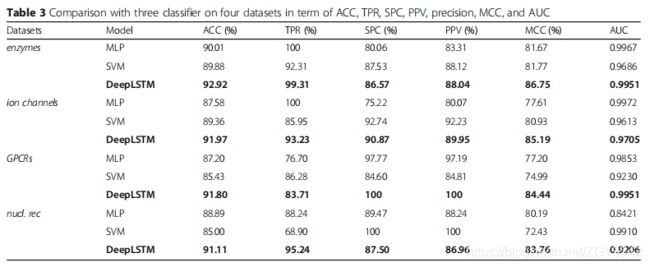
4.2 与最先进的方法进行比较
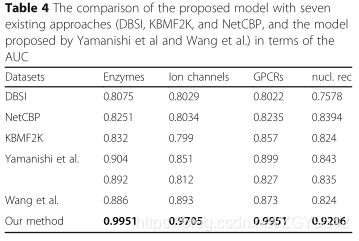
5.讨论
在本文中,作者开发了一种基于深度学习的方法,可以使用化合物和蛋白质序列来推断潜在的DTI。为了评估提出方法的能力,将其与几种最新方法进行了比较。实验结果证明,该方法在性能方面明显优于其他方法。与其他分类器相比,提供了初步证据,表明DeepLSTM在DTI任务上优于传统机器学习系统。通过使用SPCA融合PubSM指纹和PSSM和LM组合获得的蛋白质进化特征用于药物靶标的表征和定量方法,提出了一个有趣的方案。当表征方法分别与三个不同的分类器配合使用时,观察到有希望的结果。这些结果表明,该方案在特征表达和识别方面具有很大的优势。作者已经证明,所提出的方法可以与较小的数据集配合使用,这与以前的方法有所不同,并且以其自身的特殊方式进行。我们还发现,随着数据集大小的增加,预测质量继续提高。这强调了该模型用于训练和应用非常大的数据集的价值,并建议通过增加数据大小可以进一步提高性能。总体而言,理论分析和实验结果为使用该方法预测DTI的有效性提供了有力的理论和经验证据。