CancerSubtypes包的介绍(根据生信技能树Jimmy老师分享的乳腺癌分子分型包资料整理)
CancerSubtypes包的介绍(根据生信技能树Jimmy老师分享的乳腺癌分子分型包资料整理,感谢Jimmy老师!)
- 1. 引言
- 2. 数据处理
-
- 2.1 基本处理
-
- 2.1.1 通过检查数据分布来分析原始数据
- 2.1.2 具有缺失值(NA)特征的数据插补
- 2.1.3 数据标准化
- 2.2 特征选择
-
- 2.2.1 基于最大方差的特征选择
- 2.2.2 基于最大方差的绝对中位差的特征选择
- 2.2.3 基于主成分分析的特征降维和提取
- 2.2.4 基于Cox回归模型的特征选择
- 3. 用于癌症亚型识别的聚类方法
-
- 3.1 用于癌症亚型识别的共识聚类
- 3.2 用于癌症亚型识别的共识非负矩阵分解
- 3.3 用于癌症亚型识别的综合聚类
- 3.4 用于癌症亚型识别的相似性网络融合
- 3.5 用于癌症亚型识别的SNF、CC结合方法
- 3.6 用于癌症亚型识别的加权相似性网络融合
- 4. 已鉴定癌症亚型的结果验证、解释和可视化
-
- 4.1 轮廓宽度
- 4.2 生存分析
- 4.3 聚类的统计显著性
- 4.4 差异表达分析
期刊: Bioinformatics
论文: CancerSubtypes: an R/Bioconductor package for molecular cancer subtype identification, validation and visualization
Github link: https://github.com/taoshengxu/CancerSubtypes/blob/master/inst/doc/CancerSubtypes-vignette.R
数据集:TCGA
亮点:R包-CancerSubtypes,用于使用 多组学数据(包括基因表达,miRNA表达和DNA甲基化数据)鉴定 癌症亚型。
CancerSubtypes包整合了四种主要的计算方法 ,这些方法在癌症亚型鉴定方面被高度引用。CancerSubtypes为数据预处理,特征选择和后续结果分析(包括结果计算、生物学验证和可视化)提供标准化框架。
每个步骤的输入和输出都以相同的数据格式打包,便于比较不同的方法。
1. 引言
CancerSubtypes是用于癌症亚型分析的软件包,包括从数据集处理到结果验证的各种功能。
在CancerSubtypes软件包中,我们提供了一个统一的框架,分析癌症亚型的原始数据以及结果可视化。主要功能包括基因组数据预处理、癌症亚型鉴定、结果验证、结果可视化和比较。
CancerSubtypes为基因组数据预处理提供了常见的数据插补和归一化方法。同时,有四种特征选择方法来筛选基因组数据集中的关键特征。常见的癌症亚型识别方法集成在此软件包中,例如共识聚类(CC)[来自R包ConsensusClusterPlus],共识非负矩阵分解(CNMF)[来自R包NMF],综合聚类(iCluster)[来自R包iCluster],相似性网络融合(SNF)[来自R包SNFtool],SNF和CC的结合方法(SNF.CC)和加权相似性网络融合 (WSNF)。
我们以统一的输入和输出数据格式应用这些癌症亚型识别方法。分析癌症亚型的过程可以在标准工作流程中轻松进行。CancerSubtypes提供了最有用的特征选择方法和亚型验证方法,帮助用户专注于他们的癌症基因组数据,并且可以轻松地以可视化的方式比较和评估不同方法的结果。
2. 数据处理
# 安装CancerSubtypes包
devtools::install_github("taoshengxu/CancerSubtypes")
# 加载TCGA数据
BiocManager::install("RTCGA")
BiocManager::install("RTCGA.mRNA") ## (85.0 MB)
library(CancerSubtypes)
library("RTCGA.mRNA")
对于基本数据处理,CancerSubtypes提供了数据分布检查、数据插补和归一化以及特征选择的方法。CancerSubtypes包中有四种特征选择方法(方差-Var,中位数绝对偏差-MAD,COX模型,主成分分析-PCA)。所有数据处理方法都具有相同的输入和输出数据格式。
2.1 基本处理
2.1.1 通过检查数据分布来分析原始数据
## 准备TCGA基因表达数据集进行分析。
rm(list = ls())
data(BRCA.mRNA)
mRNA=t(as.matrix(BRCA.mRNA[,-1]))
mRNA[1:5,1:5]
colnames(mRNA)=BRCA.mRNA[,1] #基因名×样本名
mRNA[1:5,1:5]
## 观察数据集的平均值,方差和中位数绝对偏差分布,可以帮助用户获得数据的分布特征,例如,评估数据集是否符合正态分布。
data.checkDistribution(mRNA) #平均值、方差和中位数绝对偏差分布图

2.1.2 具有缺失值(NA)特征的数据插补
原始基因组数据集始终包含缺失的观察结果,尤其是在微阵列基因表达数据中。在极少数样本中移除所有缺少观测值的特征是不明智的,因为有用的信息将被丢弃。常用方法是为缺失的观测值插补适当的值。CancerSubtypes为基因组数据集集成了三种常见的插补方法。
table(is.na(mRNA))
index=which(is.na(mRNA))
res1=data.imputation(mRNA,fun="median")
res2=data.imputation(mRNA,fun="mean")
res3=data.imputation(mRNA,fun="microarray")
2.1.3 数据标准化
result1=data.normalization(mRNA,type="feature_Median",log2=FALSE)
result2=data.normalization(mRNA,type="feature_zscore",log2=FALSE)
2.2 特征选择
2.2.1 基于最大方差的特征选择
## 选取方差最大的前 1000 个特征。
data1=FSbyVar(mRNA, cut.type="topk",value=1000)
## 选择方差>0.5的特征
data2=FSbyVar(mRNA, cut.type="cutoff",value=0.5)
2.2.2 基于最大方差的绝对中位差的特征选择
data1=FSbyMAD(mRNA, cut.type="topk",value=1000)
data2=FSbyMAD(mRNA, cut.type="cutoff",value=0.5)
2.2.3 基于主成分分析的特征降维和提取
mRNA1=data.imputation(mRNA,fun="microarray")
data1=FSbyPCA(mRNA1, PC_percent=0.9,scale = TRUE)
2.2.4 基于Cox回归模型的特征选择
data(GeneExp)
data(time)
data(status)
data1=FSbyCox(GeneExp,time,status,cutoff=0.05)
3. 用于癌症亚型识别的聚类方法
3.1 用于癌症亚型识别的共识聚类
共识聚类(CC,2003)作为一种无监督亚型识别方法,在许多基因组研究中是一种常用且有价值的方法,并且具有许多成功的应用。
##当输入数据集只有一个基因表达矩阵。
data(GeneExp)
result=ExecuteCC(clusterNum=3,d=GeneExp,maxK=10,clusterAlg="hc",distance="pearson",title="GBM")
##当输入数据集是作为列表的多基因组学数据
data(GeneExp)
data(miRNAExp)
GBM=list(GeneExp=GeneExp,miRNAExp=miRNAExp)
result=ExecuteCC(clusterNum=3,d=GBM,maxK=10,clusterAlg="hc",distance="pearson",title="GBM")
3.2 用于癌症亚型识别的共识非负矩阵分解
非负矩阵分解(CNMF,2004)作为一种有效的降维方法,用于区分高维基因组数据的分子模式,并为亚型识别提供了一种强大的方法。我们应用 NMF 包来执行癌症基因组数据集的非负矩阵分解。因此,此方法允许用户输入用于并行处理的核心 CPU 的数量。
##当输入数据集只有一个基因表达矩阵。
data(GeneExp)
result=ExecuteCNMF(GeneExp,clusterNum=3,nrun=30)
##当输入数据集是作为列表的多基因组学数据
data(GeneExp)
data(miRNAExp)
GBM=list(GeneExp=GeneExp,miRNAExp=miRNAExp)
result=ExecuteCNMF(GBM,clusterNum=3,nrun=30)
3.3 用于癌症亚型识别的综合聚类
综合聚类法(iCluster,2009)使用联合潜在变量模型对多种类型的组学数据进行综合聚类。
data(GeneExp)
data(miRNAExp)
data1=FSbyVar(GeneExp, cut.type="topk",value=1000)
data2=FSbyVar(miRNAExp, cut.type="topk",value=300)
GBM=list(GeneExp=data1,miRNAExp=data2)
result=ExecuteiCluster(datasets=GBM, k=3, lambda=list(0.44,0.33,0.28))
3.4 用于癌症亚型识别的相似性网络融合
相似性网络融合(SNF,2014)是一种用于聚合多组学数据的融合相似性网络的计算方法。
data(GeneExp)
data(miRNAExp)
GBM=list(GeneExp=GeneExp,miRNAExp=miRNAExp)
result=ExecuteSNF(GBM, clusterNum=3, K=20, alpha=0.5, t=20)
3.5 用于癌症亚型识别的SNF、CC结合方法
我们提出将SNF和CC结合在一起,以产生一种新的癌症亚型鉴定方法。
data(GeneExp)
data(miRNAExp)
data(time)
data(status)
data1=FSbyCox(GeneExp,time,status,cutoff=0.05)
data2=FSbyCox(miRNAExp,time,status,cutoff=0.05)
GBM=list(GeneExp=data1,miRNAExp=data2)
result=ExecuteSNF.CC(GBM, clusterNum=3, K=20, alpha=0.5, t=20,maxK = 10, pItem = 0.8,reps=500,
title = "GBM", plot = "png", finalLinkage ="average")
3.6 用于癌症亚型识别的加权相似性网络融合
WSNF是一种利用基因调控网络信息进行癌症亚型识别的方法。它利用miRNA-TF-mRNA调控网络来得到这些特征的重要性。
data(GeneExp)
data(miRNAExp)
data(Ranking)
##检索基因的特征排序
gene_Name=rownames(GeneExp)
index1=match(gene_Name,Ranking$mRNA_TF_miRNA.v21_SYMBOL)
gene_ranking=data.frame(gene_Name,Ranking[index1,],stringsAsFactors=FALSE)
index2=which(is.na(gene_ranking$ranking_default))
gene_ranking$ranking_default[index2]=min(gene_ranking$ranking_default,na.rm =TRUE)
##检索基因的特征排序
miRNA_ID=rownames(miRNAExp)
index3=match(miRNA_ID,Ranking$mRNA_TF_miRNA_ID)
miRNA_ranking=data.frame(miRNA_ID,Ranking[index3,],stringsAsFactors=FALSE)
index4=which(is.na(miRNA_ranking$ranking_default))
miRNA_ranking$ranking_default[index4]=min(miRNA_ranking$ranking_default,na.rm =TRUE)
##聚类
ranking1=list(gene_ranking$ranking_default ,miRNA_ranking$ranking_default)
GBM=list(GeneExp,miRNAExp)
result=ExecuteWSNF(datasets=GBM, feature_ranking=ranking1, beta = 0.8, clusterNum=3,
K = 20,alpha = 0.5, t = 20, plot = TRUE)
4. 已鉴定癌症亚型的结果验证、解释和可视化
通过计算方法鉴定出的癌症亚型应符合生物学意义,并揭示明显的分子模式。
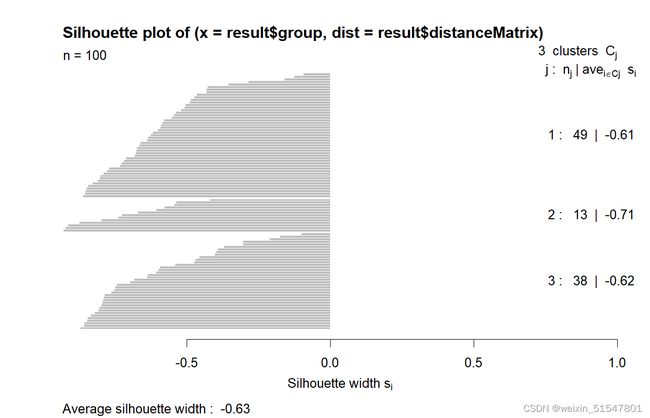
4.1 轮廓宽度
轮廓宽度用于评价与其他亚型相比,样本与其被识别的亚型的匹配程度,值越高表示匹配越好。每条水平线表示轮廓图中的一个样本,线长为样本的轮廓宽度。
data(GeneExp)
data(miRNAExp)
GBM=list(GeneExp=GeneExp,miRNAExp=miRNAExp)
result=ExecuteSNF(GBM, clusterNum=3, K=20, alpha=0.5, t=20,plot = FALSE)
##相似性矩阵
sil=silhouette_SimilarityMatrix(result$group, result$distanceMatrix)
plot(sil)

注:如果输入矩阵是样本之间的差异矩阵,请使用聚类包中的 silhouette() 来计算轮廓宽度,否则会产生错误的结果。
sil1=silhouette(result$group, result$distanceMatrix)
plot(sil1) ##错误的结果 #所有样本的轮廓宽度均为负数。
4.2 生存分析
生存分析用于判断亚型之间的不同生存模式。
data(GeneExp)
data(miRNAExp)
data(time)
data(status)
data1=FSbyCox(GeneExp,time,status,cutoff=0.05)
data2=FSbyCox(miRNAExp,time,status,cutoff=0.05)
GBM=list(GeneExp=data1,miRNAExp=data2)
## 1.ExecuteSNF
result1=ExecuteSNF(GBM, clusterNum=3, K=20, alpha=0.5, t=20,plot = FALSE)
group1=result1$group
distanceMatrix1=result1$distanceMatrix
p_value=survAnalysis(mainTitle="GBM1",time,status,group1,
distanceMatrix1,similarity=TRUE)
##
## *****************************************************
## GBM1 Cluster= 3 Call:
## survdiff(formula = Surv(time, status) ~ group)
##
## N Observed Expected (O-E)^2/E (O-E)^2/V
## group=1 46 44 41.3 0.173 0.311
## group=2 40 39 27.3 4.990 7.387
## group=3 14 13 27.4 7.529 11.661
##
## Chisq= 14.2 on 2 degrees of freedom, p= 8e-04
这是一个由三部分组成的组合图:生存曲线、样本相似性矩阵热图和已识别癌症亚型的轮廓宽度图。所有图中的样本都已按识别的癌症亚型进行了重组。这种图形提供了易于评估的可见结果。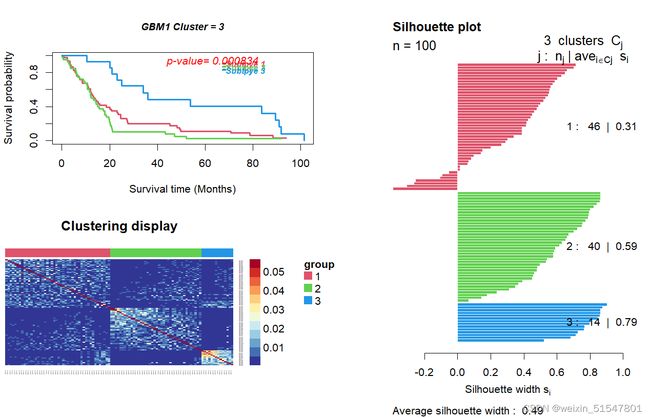
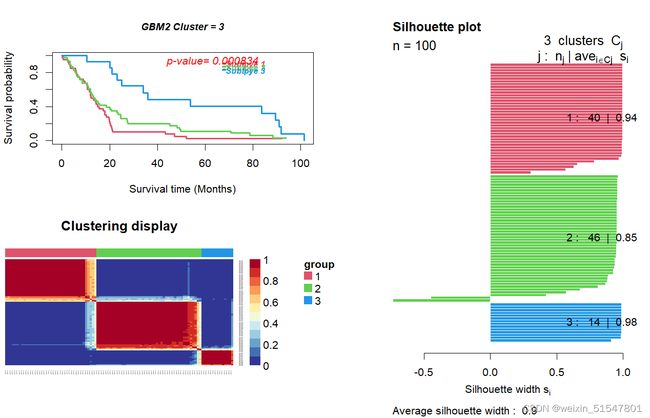
## 2.ExecuteSNF.CC
result2=ExecuteSNF.CC(GBM, clusterNum=3, K=20, alpha=0.5, t=20,
maxK = 5, pItem = 0.8,reps=500,
title = "GBM2", plot = "png",
finalLinkage ="average")
group2=result2$group
distanceMatrix2=result2$distanceMatrix
p_value=survAnalysis(mainTitle="GBM2",time,status,group2,
distanceMatrix2,similarity=TRUE)
##
## *****************************************************
## GBM2 Cluster= 3 Call:
## survdiff(formula = Surv(time, status) ~ group)
##
## N Observed Expected (O-E)^2/E (O-E)^2/V
## group=1 40 39 27.3 4.990 7.387
## group=2 46 44 41.3 0.173 0.311
## group=3 14 13 27.4 7.529 11.661
##
## Chisq= 14.2 on 2 degrees of freedom, p= 8e-04
4.3 聚类的统计显著性
聚类的统计显著性是一种纯粹的统计方法,用于检验亚型之间的显著性差异数据分布。不同的表达式测试每个亚型和一个参考组(一组正常样本)之间的表达差异。
data(GeneExp)
data(miRNAExp)
data(time)
data(status)
GBM=list(GeneExp=GeneExp,miRNAExp=miRNAExp)
result=ExecuteSNF(GBM, clusterNum=3, K=20, alpha=0.5, t=20,plot = FALSE)
group=result$group
sigclust=sigclustTest(miRNAExp,group, nsim=1000, nrep=1, icovest=1)
sigclust
# Subtype 1 Subtype 2 Subtype 3
# Subtype 1 1.000 0.018 0.000
# Subtype 2 0.018 1.000 0.234
# Subtype 3 0.000 0.234 1.000
补充材料中SigClust 汇总图显示了聚类索引 (CI) 的分布。代表模拟CI的蓝点使用随机垂直抖动绘制,以获得更好的可视化效果。实线和虚线对应于拟合模拟CI的估计非参数密度和高斯密度。p 值显示两个子类型之间的有效水平。

4.4 差异表达分析
差异表达分析是测试每个亚型和参考组(一组正常样本)之间的表达差异。在这里,我们应用limma包在每个亚型和正常样本之间进行不同的表达分析。
library("RTCGA.mRNA")
#require(TCGAbiolinks)
rm(list = ls())
data(BRCA.mRNA)
mRNA=t(as.matrix(BRCA.mRNA[,-1]))
colnames(mRNA)=BRCA.mRNA[,1]
mRNA1=data.imputation(mRNA,fun="microarray")
mRNA1=FSbyMAD(mRNA1, cut.type="topk",value=5000)
## 将正常和肿瘤样本分开
index=which(as.numeric(substr(colnames(mRNA1),14,15))>9)
mRNA_normal=mRNA1[,index]
mRNA_tumor=mRNA1[,-index]
## 移除重复样本
index1=which(as.numeric(substr(colnames(mRNA_tumor),14,15))>1)
mRNA_tumor=mRNA_tumor[,-index1]
## 识别癌症亚型
result=ExecuteCC(clusterNum=5,d=mRNA_tumor,maxK=5,clusterAlg="hc",distance="pearson",title="BRCA")
group=result$group
res=DiffExp.limma(Tumor_Data=mRNA_tumor,Normal_Data=mRNA_normal,group=group,topk=NULL,RNAseq=FALSE)
## subtype 1中的差异表达基因
head(res[[1]])
## ID logFC AveExpr t P.Value adj.P.Val
## 2439 COL10A1 4.712350 5.0043839 38.57960 3.234769e-151 1.617385e-147
## 3725 MMP11 3.528733 2.6563911 34.61589 3.206291e-134 8.015728e-131
## 3797 CA4 -4.357770 -1.0831256 -27.32458 7.230942e-101 1.205157e-97
## 3235 CAV1 -3.006418 -1.5612746 -26.90504 7.051163e-99 8.813954e-96
## 4586 CXCL3 -2.499337 -2.9294699 -26.24577 9.675861e-96 9.675861e-93
## 4133 RYR3 -2.575299 -0.4590268 -25.94906 2.524404e-94 2.103670e-91
## B
## 2439 334.8796
## 3725 295.9708
## 3797 219.5287
## 3235 214.9674
## 4586 207.7722
## 4133 204.5236
## subtype 2中的差异表达基因
head(res[[2]])
## ID logFC AveExpr t P.Value adj.P.Val B
## 637 ITM2A -6.173926 0.8088468 -12.356546 1.091109e-18 5.455546e-15 31.11131
## 4942 CKAP2 4.229372 -2.1539566 9.959475 1.093416e-14 2.267182e-11 22.64778
## 4596 RCBTB2 -4.492770 -0.2484435 -9.904508 1.360309e-14 2.267182e-11 22.44505
## 3297 GPR19 4.272492 -2.2195806 9.791601 2.132269e-14 2.665337e-11 22.02758
## 4829 CASP4 -3.767096 0.7556694 -9.613507 4.342293e-14 4.342293e-11 21.36631
## 2065 MCM4 5.079992 -3.2200565 9.518389 6.355510e-14 5.296258e-11 21.01180