scMDC:针对单细胞多模态数据进行聚类

目录
- 摘要
- 引言
-
- 背景介绍
- 单细胞数据聚类方法回顾
- ZINB
- scMDC
摘要
单细胞多模态测序技术的发展是为了在同一细胞中同时分析不同模态的数据,它为在单细胞水平上联合分析多模态数据从而识别不同细胞类型提供了一个独特的机会。正确的聚类结果对于下游复杂生物功能研究至关重要。然而,结合不同模态数据进行聚类分析仍然是一个统计学和计算上的挑战。为此,作者提出了一种新的多模态深度学习方法scMDC,用于单细胞多组学数据聚类分析。大量的模拟数据和真实数据实验表明,scMDC在不同的单细胞多模态数据集上均优于现有的单细胞单模态和多模态聚类方法。此外,运行时间的线性可扩展性使scMDC成为分析大型多模态数据集的有效方法。
引言
背景介绍
单细胞RNA测序(scRNA-seq)可获取RNA模态下单个细胞内的高分辨率图谱。基于scRNA-seq技术,近年来又开发了多种多模态测序技术,以联合分析单个细胞中的多模态数据。例如,CITE-seq和REAP-seq用于在细胞水平上测量mRNA表达和表面蛋白丰度。除了研究单细胞转录组和表面蛋白外,scATAC-seq的发展提供了测量单细胞染色质可及性的机会。当然,现在也已经开发的一些多组学单细胞测量技术,可以联合测量单细胞内的染色质可及性和基因表达,例如SNARE-seq和10X单细胞多组学ATAC +基因表达(Single-Cell Multiome ATAC + Gene Expression,SMAGE-seq)。总之,这些多模态测序技术提供了更全面的单细胞图谱。
在多模态数据中,不同模态提供的生物学信息是互补的。以CITE-seq为例,其ADT模态聚焦于表面蛋白丰度。ADT数据往往具有较低的丢失率(dropout rate),因此可以可靠地量化细胞活性。对于scMDC研究分析的五个CITE-seq数据集,其ADT数据的丢失率仅有12%。相比之下,其相应的mRNA数据中有超过80%甚至90%的条目为零。对于大多数基因来说,蛋白质是实现其功能的最终产物,mRNA是其直接产物。因此,ADT数据似乎是表征细胞功能和类型的理想方法。然而,由于目前技术的限制,ADT只能分析数百种蛋白质。因此,ADT数据善于识别常见的细胞类型。然而,由于ADT数据的维度有限,它可能不能很好地检测罕见细胞类型。相比之下,mRNA数据的全转录组可以捕获全面的细胞类型。然而,基于scRNA-seq的细胞聚类可能会受到高丢失率(高维稀疏性)的影响。此外,在考虑转录和翻译时,同一基因产生的ADT和mRNA来源的数量并不相同。在这种情况下,ADT和mRNA数据为细胞类型识别提供了互补信息。SNARE-seq、SMAGE-seq以及scATAC-seq数据提供的染色质可及性信息也是mRNA数据的补充。因此,通过整合来自多模态的信息,能够获得更高分辨率的细胞分型。
单细胞数据聚类方法回顾
传统方法不适用于多模态数据聚类:聚类分析是大多数单细胞研究中必不可少的步骤,并且已经得到了广泛的研究。基于聚类结果,研究人员可以探索细胞类型或亚型水平的生物活性。目前已经设计了许多聚类方法来分析scRNA-seq数据,例如Tscan、 Seurat和SC3。然而,这些传统的单细胞聚类方法不能充分利用多组学数据的优势来提高聚类性能,因此不适用于多模态数据。
对CITE-seq的聚类:过去几年中,出现了几种用于CITE-seq数据的聚类分析方法。作者也在最近提出了一种单细胞深度约束聚类框架scDCC,该框架可以通过手动定义的约束将ADT信息集成到scRNA-seq数据的聚类分析中。BREM-SC是一种分层贝叶斯混合模型,采用两个多项式模型联合表征scRNA-seq和ADT数据。它假设多项式模型中的比例(基因或蛋白质的相对表达水平)遵循狄利克雷分布,并引入细胞特异性随机效应来模拟两个数据源之间的相关性。虽然BREM-SC是最早提出的用于CITE-seq数据聚类分析的模型之一,但它有几个局限性。首先,它假设数据遵循特定的分布,但这种参数假设可能并不适用于所有的实际应用。其次,BREM-SC并不能表征dropout事件,这是scRNA-seq数据聚类的主要问题。最后,BREM-SC存在可扩展性问题。当分析数千个细胞时,BREM-SC的运行时间大大增加。
同时,CiteFuse、Seurat V4和Specter可以使用基于距离的图来聚类CITE-seq数据。CiteFuse分别计算ADT和mRNA的细胞间相似性矩阵,再通过相似性网络融合算法将两者合并,之后采用spectral和Louvain算法等基于图的聚类算法对合并后的相似性矩阵进行聚类。然而,基于相似性矩阵的聚类不能明确考虑scRNA-seq数据中的dropout事件。Seurat V4开发了一种用于多组学数据聚类的加权最近邻(WNN)程序。WNN学习多模态数据的权重,并通过mRNA和蛋白质视图的加权组合生成细胞的相似性图。Spector是一种谱聚类方法,用于聚类具有线性时间可扩展性的单细胞数据。与BREM-SC和CiteFuse算法相比,WNN算法和Specter算法运行速度更快,占用内存更少。但是,这两种方法也没有考虑到数据中的dropout事件。
先学习联合embedding再聚类:另一项相关的研究方向是学习不同模态的联合嵌入,这种联合嵌入有望改进包括聚类在内的各种下游分析。TotalVI是一种深度变分自编码器,可以捕获不同数据类型的相同潜在空间。通过这种设计,TotalVI可以从CITE-seq数据中学习配对ADT和mRNA测量的联合概率表示,这些数据解释了每种模态的不同信息。同样,对于SNARE-seq或SMAGE-seq数据,Cobolt和scMM采用多模态变分自编码器对多种模态进行联合建模,并学习单细胞mRNA-seq和ATAC-seq数据的联合嵌入。然而,这些专注于联合嵌入的方法并不是为聚类而设计和优化的,其划分策略对于聚类来说不是最优的。
考虑dropout事件:综上可知,在学习联合嵌入以及聚类的过程中,现有的很多方法都没有考虑到单细胞数据中的dropout事件。然而,普遍存在的dropout事件使得单细胞计数数据存在零膨胀和过度分散的问题。为了更好地表征单细胞mRNA计数数据,零膨胀负二项(ZINB)模型被广泛用于解释dropout事件。许多基于ZINB模型的方法(包括深度学习方法)已被开发用于分析scRNA-seq计数数据,其中包括ZINB- WaVE、DCA、scVI和scDeepCluster。这些研究表明,ZINB模型可以有效地表征scRNA-seq数据,并提高表征学习和聚类结果。
ZINB
ZINB可以回顾DCA(Single-cell RNA-seq denoising using a deep count autoencoder),DCA研究的是Single-cell RNA sequencing denoising,也就是单细胞RNA测序的去噪,由于dropout问题,会干扰scRNA-seq的数据分析,因此需要有降噪技术用于稀疏的scRNA-seq数据,因此DCA通过negative binomial noise model with or without zero-inflation对数据进行建模。
在多数scRNA-seq的研究中,会经常看到描述scRNA-seq数据服从负二项分布(Negative Binomial, NB)。因为:
- 负二项分布算是最接近scRNA-seq的数据分布。scRNA-seq数据的离散通常是高度扭曲的,方差往往会大于均值,因此不适合采用泊松分布来近似(泊松分布的均值和方差是相等的),而通常scRNA-seq的数据的方差会随着均值的增大而更加增大,比如下面这个图:
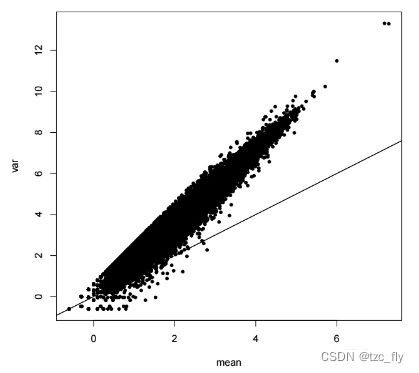
- 随着均值的增大,scRNA-seq的数据会越来越偏离泊松分布的直线,方差会大于均值,也就是over-dispersion现象(过度离散化),因此近年来采用负二项分布去近似scRNA-seq的数据(NB的方差和均值是二次函数关系)。
ZINB描述为,任取 π ∈ ( 0 , 1 ) \pi\in(0,1) π∈(0,1), δ 0 ( x ) \delta_{0}(x) δ0(x)为0点处的脉冲函数: Z I N B ( x ; π , μ , θ ) = π δ 0 ( x ) + ( 1 − π ) N B ( x ; μ , θ ) ZINB(x;\pi,\mu,\theta)=\pi\delta_{0}(x)+(1-\pi)NB(x;\mu,\theta) ZINB(x;π,μ,θ)=πδ0(x)+(1−π)NB(x;μ,θ)ZINB就是在NB的基础上,在0点处增加了一个脉冲函数,ZINB是两者的加权, 第一项可以看做是整个数据找那个0出现的次数(密度函数就是单独一个点的值为1),所以ZINB可以捕捉数据中的高稀疏性,可以准确纠正0点噪声。同时,作为输入数据,同一个batch的数据还进行了归一化。
ZINB loss有3个重要参数, π \pi π代表脉冲函数的权重,即dropout的比例, μ \mu μ代表NB的均值, θ \theta θ代表分布的稀疏程度,DCA希望通过AE结构学习三个参数的分布,而不仅仅是数据的去噪和恢复。
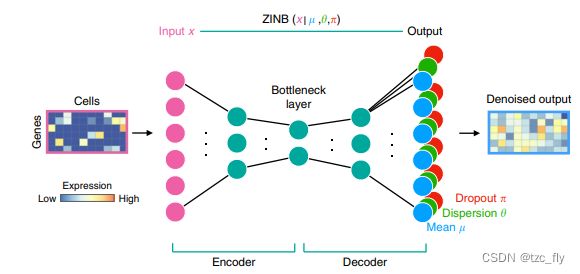
- DCA架构,最左边的cells是输入,将每个细胞按照向量的形式输入,其中深蓝色的代表0,颜色越浅,表示基因表达度越高,然后输入到图中粉红色的节点,中间的结构和AE相同,但是最后一层和传统的AE不一样,同时有三层输出,分别代表ZINB的三个隐藏参数,用了蓝绿红三种颜色表示,蓝色即均值表示去噪后的数据。
scMDC
在该论文中,作者提出了一种用于多模态单细胞数据聚类分析的多模态深度学习模型scMDC,scMDC的网络架构如图1所示。scMDC采用多模态自动编码器,对不同模态的concat数据采用一个编码器,对每个模态的数据分别采用两个解码器进行解码。在scDeepCluster之后,使用ZINB损失作为重建损失,并将瓶颈层用于深度k均值聚类。为了进一步改进潜在特征学习,作者引入了一种基于Kullback-Leibler散度的损失(KL loss),该损失吸引相似的细胞,分离不同的细胞。scMDC同时进行优化,包括自动编码器、KL loss和深度k均值聚类。scMDC是一种端到端的多模态深度学习聚类方法,用于建模不同的多组学数据。利用GPU的优势,scMDC在大型数据集的分析中非常高效。此外,通过使用条件自动编码器框架,scMDC可以在分析多批次数据时校正批次效应。scMDC是第一个端到端的深度聚类方法,既可以集成多模态数据,又可以消除不同类型多模态数据的批次效应。从对CITE-seq和SMAGE-seq数据的大量实验中可以观察到scMDC的优越性能。聚类后,对于给定的簇,作者还通过将ACE模型(ACE: explaining cluster from an adversarial perspective)移植到scMDC中来检测标记物(基因或蛋白质),并根据从ACE学习到的基因排序进行基因集富集分析。这些下游分析的结果又进一步证明了scMDC聚类性能的优越性。

- 图1a:scMDC具有用于级联数据的一个编码器和用于多模态数据的每个模态的解码器。它可以用于聚类CITE-seq数据和SMAGE-seq数据。螺旋符号表示添加到数据中的人工噪声。对于多批次数据集,scMDC将以条件自动编码器的方式工作。one-hot batch vector B(b维)将连接到编码器(具有原始特征维度m)和解码器(具有潜在特征维度z)的输入特征。这是为批次校正设计的。scMDC学习latent表征Z(具有潜在特征维度z),其集成了不同模态的信息。在Z上执行深度k均值算法和KL loss。
- 图1b:基于聚类结果,scMDC使用ACE模型来检测不同聚类中的marker。
- 图1c:然后,可以基于ACE学习的基因等级进行通路分析。
关于下游分析
基于聚类结果,作者进行了两种流行的下游分析,差异表达分析(DE)和基因集富集分析(GSEA)。作者采用ACE算法,根据基因的置信度对基因进行排序,并将其分配到一个簇中。DE分析可以在两个簇之间进行,也可以在一个簇和其他簇之间进行。然后,根据归一化的mRNA计数计算每个基因的对数变化,得到差异表达的方向。通过基因排序和方向,再进行GSEA来找到目标簇中的富集通路。在本文中,作者对BMNC数据中的四个最大集群进行了DE和GSEA,所有比较都是在目标簇和其他簇之间进行的。这些下游分析进一步巩固了scMDC聚类结果的正确性。
- 基因集富集分析(Gene Set Enrichment Analysis,GSEA)是一种计算方法,用来确定一组先验定义的基因集是否在两种生物状态之间显示出统计学上显著的、一致的差异。