NeurIPS 2022 | 当AI遇上量子化学:腾讯Al Lab冠军模型提升27%
感谢阅读腾讯 AI Lab 微信号第 159 篇文章。本文为腾讯 AI Lab 在 NeurIPS 2022 第二届 Open Catalyst Challenge (OCP)竞赛中夺冠的解决方案解析。
在刚刚落幕的由 Meta AI 研究院及卡耐基梅隆大学(CMU)联合机器学习顶级会议 NeurIPS 共同举办的第二届 Open Catalyst Challenge (OCP)竞赛中,由腾讯 AI Lab 领头,中国人民大学,清华大学以及香港中文大学组成的联合团队 TTRC 以 0.396eV 绝对误差的成绩获得第一,相对于去年的最好成绩,提升达到 27.6%。
比赛链接:https://opencatalystproject.org/challenge.html
项目主页:https://ai.tencent.com/ailab/ml/ocp/index.html
本文为具体技术方案解析。
背景
Open Catalyst Project(OCP)是由 Meta AI 和卡耐基梅隆大学联合发起的一个科研项目,旨在使用人工智能算法加速可用于再生能源存储的催化剂的发现。其核心目的是通过寻找高效且经济的催化剂来解决再生能源存储的问题。
在这一领域,传统方法是基于量子力学(密度泛函理论 DFT)的模拟计算催化剂表面和目标吸附物的结合能来测试和评估新的催化剂结构。然而,这类模拟的一大缺点是其巨大的计算成本,通常单个组合模拟就需要耗费 24 小时以上。这使得我们无法高效且大量地筛选潜在的催化剂结构。因此,如何利用机器学习算法去建模量子化学模型进而实现对于催化剂结构的有效筛选这是一个非常具有前景的方向。
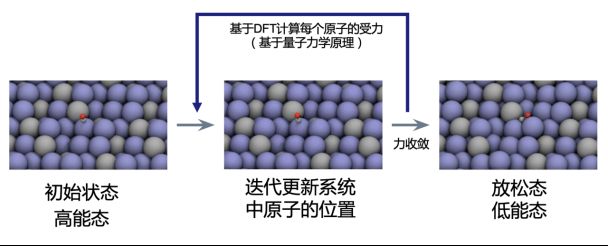
传统基于量子力学的计算流程(引自官方资料)
给定一个催化剂-吸附物组成的反应系统,经典的量子力学模拟计算分为两个步骤,第一步是基于密度泛函理论计算系统中微观粒子的等效的受力。第二步则是基于这个受力迭代更新系统中原子核的位置。直到受力收敛。这样就可以得到这个系统低能态,即松弛状态,进而计算催化剂和吸附物的结合能。
本次比赛参赛主题则是构造机器学习模型预测一个由催化剂-吸附物组成的反应系统的松弛状态能量,即 IS2RE(Initial state to relaxed energy)。同上一届不同的是,除了提供催化剂-吸附物以及对应能量标签(46万)外,主办方还额外提供了200万的由密度泛函计算得到的静态反应系统的坐标及其对应的力和能量。基于这些数据,模型可以去预测静态结构下的等效受力,即 S2EF(Structure to energy and force),使得模型可以更好探索量子力学计算的部分的建模。
技术分析
自从 OCP 项目发起以来,由于其重要的科学意义和庞大的数据规模以及具有挑战的任务,吸引了 DeepMind,MSRA,达摩院,MILA,MIT,上海交大等机构的关注和参与。在今年的第二届比赛中,我们提出的 GeoEnsemble 框架以 0.396eV 绝对误差的成绩获得第一,相对于去年的比赛的最好成绩Graphormer,提升达到 27.6%。
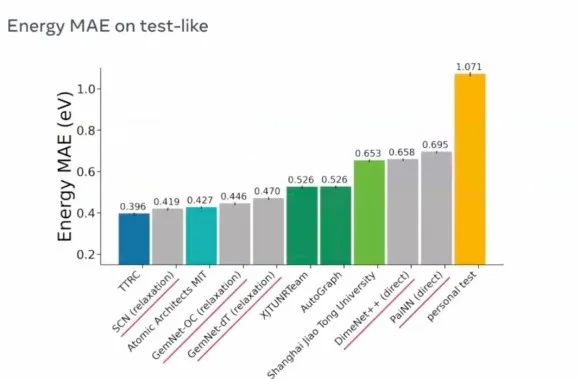
非公开测试集排行榜,带下划线的为官方Baseline。(引自官方资料)

本届比赛的方案相对于上一届方案的提升。(引自官方资料)

限定比赛用训练集的方法,在Public Leaderboard性能表现。
数据来源: https://eval.ai/web/challenges/challenge-page/712/leaderboard/1950
在 GeoEnsemble 框架中,为了对原子之间复杂的动态交互关系进行建模,
我们在 ICLR 2022 发表的图动力学神经网络(GMN)[1] 的基础上进行了改进,提出了 GMN-OC 模型。
GMN-OC 模型的输入是一个由原子构成的几何图,几何图中包含了几何特征(原子的三维坐标)和非几何特征(原子的类型),模型可以预测输出几何向量(原子的受力)和非几何标量(系统能量)。

GMN-OC整体输入和输出流程
在 GMN-OC 模型中,我们构造了一个基于多通道的几何特征O(3)等变函数 与不变函数用来处理几何特征和非几何特征的交互。
基于这两个函数,我们构建了一个基于消息传递的图神经网络,在 GMN-OC 的每一个网络层中,会基于O(3)等变与不变函数进行消息传递与聚合,从而实现对每个原子的几何特征和非几何特征进行更新。
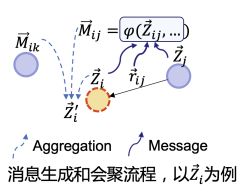
基于消息传播的几何特征更新过程
在这个基础模型上,我们进一步引入了一个全局共享表示模块(Global Representation Module)建模在 DFT 计算中可能涉及到的粒子之间的全局交互信息。同时也保持了模型的等变性。

全局共享表示模块示意图
整个 GMN-OC 模型结构如下图。同时,我们在计算时,使用了 Multi-head 的显存优化方法,使得模型可以更好地应对大数据的处理。
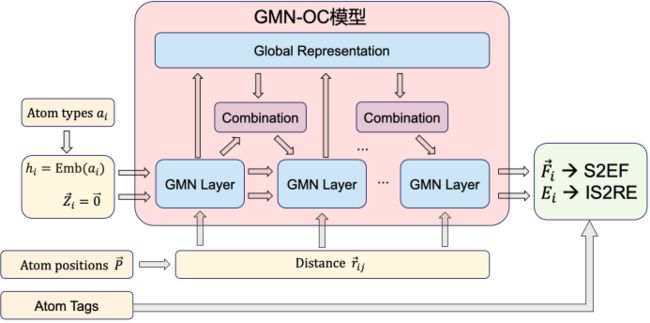
GMN-OC 模型整体架构
此外在训练技术上,为了充分利用赛事提供的两组训练数据,我们使用了 Pretrain-Finetune、Multi-task Learning 等优化技术,进一步提高模型预测精度。此外,我们结合 GMN-OC / SCN / GemNet 等多个模型,构建了多模型融合方案 GeoEnsemble。为了提高模型训练速度,我们还使用了混合精度训练等技术,并在大规模集群上完成了分布式训练。

GeoEnsemble 在训练上的改进
未来展望
利用人工智能技术助力自然科学领域中的探索和发现,已经成为近年来人工智能领域备受瞩目的应用方向之一。得益于高性能计算能力和前沿人工智能技术的不断发展,基础科学领域的研究者得以利用人工智能算法去加速相关领域中复杂、大规模的计算和模拟任务,如蛋白质折叠、小分子结合能计算、催化剂发现等。
腾讯 AI Lab 基于在人工智能算法研究中的积累,在生命科学,物理建模等多个应用方向上取得了重要的突破。例如首个大规模小分子预训练模型 GROVER [2]及骨架跃迁生成模型[3],自研蛋白质折叠模型 tFold [3]和基于序列预训练的抗体结构预测模型 tFold-Ab [4],基于等变性的蛋白质对接模型 EquiDock [5],基于图动力学网络的蛋白质动态结构预测模型 EGHN [6]等。同时,腾讯云深AI药物发现平台研发的基于等变图神经网络的分子能量框架 DeepQC,可以实现对类药分子的高精度的量化计算。
我们将在近期开源这次比赛使用的模型和训练推理源码,以助力人工智能在量子化学模拟和电子结构计算等基础研究领域的应用。在未来,腾讯 AI Lab 将持续研发和落地 AI 新技术,推动 AI 在交叉学科中的新应用,探索 AI 赋能科学发现的新范式。
参考链接:
1. Equivariant Graph Mechanics Networks with Constraints
2. Self-Supervised Graph Transformer on Large-Scale Molecular Data
3. A novel scalarized scaffold hopping algorithm with graph-based variational autoencoder for discovery of JAK1 inhibitors
4. When homologous sequences meet structural decoys: Accurate contact prediction by tFold in CASP14—(tFold for CASP14 contact prediction)
5. tFold-Ab: Fast and Accurate Antibody Structure Prediction without Sequence Homologs
6. Independent SE(3)-Equivariant Models for End-to-End Rigid Protein Docking
7. Equivariant Graph Hierarchy-Based Neural Networks

* 欢迎转载,请注明来自腾讯AI Lab微信(tencent_ailab)