iMeta | 复杂热图(ComplexHeatmap)可视化文章最新版,画热图就引它
点击蓝字 关注我们
复杂热图可视化

https://doi.org/10.1002/imt2.43
PROTOCOL
●2022年8月,德国癌症研究中心顾祖光在iMeta在线发表了题为“Complex heatmap visualization”的方法类文章。
● 该研究系统性地介绍了 ComplexHeatmap R包在复杂热图可视化方面的特性和功能。
● 第一/通讯作者:顾祖光 ([email protected])

文章上线仅3个月,全文下载近万次(iMeta下载量最高论文),Twitter上点赞过千,被引7次。
引文格式:Zuguang Gu. 2022. Complex heatmap visualization. iMeta 1: e43. https://doi.org/10.1002/imt2.43
摘 要
热图是一种广泛使用的针对矩阵数据的统计可视化方法,其用于揭示存在于矩阵中的相似模式。在 R 编程语言中,有许多绘制热图的包。其中,ComplexHeatmap 软件包为构建高度可定制的热图提供了最丰富的工具集。ComplexHeatmap 可以通过自动拼接和调整多个热图以及添加复杂注释,轻松建立多个来源信息之间的关联,因此 ComplexHeatmap 被广泛应用于许多领域的数据分析中,特别是生物信息学,以发现隐藏在数据中的关键结构。在本文中,我们全面介绍了 ComplexHeatmap 的现状,包括模块化设计、丰富的功能和广泛的应用。
关键词:热图,可视化,聚类,Bioconductor,R软件包
亮 点
● 复杂热图是一种用来揭示多种信息之间复杂关联的强大的可视化方法;
● 我们开发了一个名为 ComplexHeatmap 的 R 软件包,其中提供了大量的热图可视化工具,在生物信息学领域中被广泛使用;
● 在这篇论文中,我们系统性地介绍了 ComplexHeatmap 软件包当前的特性和功能。
视频解读
Bilibili:https://www.bilibili.com/video/BV1AW4y117kU
Youtube:https://youtu.be/Y6l29nUIrAs
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
热图是一种广泛使用的通过将颜色作为主要图形元素来可视化矩阵数据的方法。热图可视化有两大类:空间热图(spatial heatmap)和网格热图(grid heatmap) [1]。第一类可视化数据在空间分布的模式,例如全球温度分布,或用户在网页上的点击活动。一种被称为等值线图(choropleth map) 的图形使用热图来可视化地理区域的某些统计特征。第二类热图只是使用颜色作为图形元素的二维矩形布局,其中两个维度分别对应两种类型的变量。在大多数情况下,我们使用一些方法对热图的行和列进行重新排序,以便使具有相似模式的行和列在热图上聚集在一起。大多数情况下,热图的排序主要使用层次聚类进行,因此,网格热图也被称为聚类热图(cluster heatmap)。在本文中,我们只讨论网格热图。
热图可视化可以追溯到 19 世纪,当时它被用于可视化巴黎不同地区的各种社会学统计指标 [2]。然而,作为一种统计可视化方法,直到 1990 年代应用于生物信息学研究之后才被广泛使用。自从 1998 年发表的一篇关于基因表达数据热图可视化的早期论文 [3] 以来,热图一直是可视化各种组学数据的标准工具,例如基因表达谱或 DNA 甲基化数据。如今,热图也被应用于基因组学的各种研究,例如,可视化三维 (3D) 尺度上的基因组水平调控 [4]、基因组水平的 DNA 甲基化信号 [5],或围绕特定基因组区间的基因组信号的分布模式 [6]。矩形布局是最常用的热图可视化的布局方式,此外,热图还有其他布局,如圆形布局 [7]、螺线布局 [8] 和希尔伯特曲线布局 [9]。它们在特定场景中很有用。
R 是一种流行的用于数据分析和可视化的编程语言。在 R 中,有许多制作热图的软件包。stats 包中的 heatmap() 函数提供了最基本的但是很有限的功能。gplots 包中的 heatmap.2() 函数是 heatmap() 的增强版本,它支持在热图上添加更多图形,例如具有数据分布的图例,和显示与列或行的中位数之间的差异。ggplot2 包 [10] 中的 geom_tile() 函数也提供了热图的简单实现。还有一些包能够对热图提供更灵活的控制,例如 pheatmap 包中的 pheatmap() 函数和 NMF 包中的 aheatmap() 函数 [11]。
随着数据在大小和维度上迅速增长,特别是在基因组学领域,目前迫切需要一种用于集成分析或者多组学分析的有效的可视化工具用来关联多种类型的数据,以便轻松揭示多种数据之间的关系。从热图可视化的角度来看,可以体现在如下两点。第一是热图注释的支持。热图注释包含了与主热图相关联的额外信息。例如,在典型的对基因表达数据的热图可视化中,热图的行对应着基因,列对应着病人。病人通常具有其他的临床元数据,例如年龄、性别或病人是否具有某些 DNA 突变。通过附加到热图上的注释,很容易识别,例如,一组表现出高表达的基因是否与某个年龄间隔相关,或者它们是否具有特定类型的 DNA 突变。heatmap() 和 heatmap.2() 仅支持一个数字型或字符型向量的单个注释。pheatmap() 和 aheamtap() 允许多个注释。superheat [12] 和 heatmap3 [13] 包支持更多类型的注释图形,例如点或者线,通过这些额外支持的注释图形能够对数据进行更准确的视觉映射。第二点是通过同时集成多个热图直接实现“复杂热图可视化”,从而可以直接比较热图之间的相关模式。例如,在我们之前的研究 [6] 中,我们将复杂热图可视化应用于基因表达、DNA 甲基化和各种组蛋白修饰的数据,以揭示多个人体组织之间的转录调控模式。为了实现复杂注释和热图可视化,我们开发了一个名为 ComplexHeatmap [14] 的热图包。它不仅支持其他软件包中的基本注释图形,还支持大量额外的复杂注释图形,甚至允许用户自定义自己的注释图形。ComplexHeatmap 提供了简单的语法来集成多个热图和注释,其中所有热图的行或者列会自动进行调整。其易用性和功能的全面性使得 ComplexHeatmap 被广泛应用于生物信息学。
ComplexHeatmap 项目开始于 2015 年,相应的论文于 2016 年发表 [14]。从那时起,它逐渐成为生物信息学领域的一个流行工具。它已被下载超过 50 万次,并且有其他 104 个 CRAN/Bioconductor 的软件包直接依赖于它(数据收集于 2022年6月22日)。ComplexHeatmap 已被广泛的应用在生物学研究中,例如癌症 [15]、COVID-19 [16]、单细胞 [17]、免疫学 [18] 以及其他领域例如海洋学 [19]和生态学 [20]等。在过去的六年中,我们一直在积极的维护ComplexHeatmap,并添加了许多新功能。我们还将文档重新编写为一本综合性书籍 (https://jokergoo.github.io/ComplexHeatmap-reference/book/)。在本文中,我们将全面介绍 ComplexHeatmap 的现状,包括其模块化设计、丰富的功能和广泛的应用。
结果与讨论
模块化设计
ComplexHeatmap 以模块化和面向对象的方式设计。ComplexHeatmap 中定义了三个主要类:Heatmap 类定义了具有多个组件的完整热图,HeatmapAnnotation 类定义了具有特定图形的热图注释,以及管理多个热图和热图注释的 HeatmapList 类。
单个热图由热图主体和各种热图组件组成(图 1A)。热图主体是具有单个颜色格子的二维排列,其中每个格子对应于输入矩阵中的一个特定值。热图组件包含标题、系统树图(dendrogram)、矩阵行和列的文字标签以及热图注释。这些组件可以放置在热图主体的四个侧面,每个组件都由为Heatmap对象定义的特定方法所管理。此外,热图主体可以在行和列上进行切分。
热图注释包含与热图的行或列相关的附加信息。ComplexHeatmap 为设置不同的注释图形和定义新的注释图形提供了丰富的支持。热图注释可以作为热图的组件放在其四个侧面,也可以独立和热图连接。HeatmapAnnotation 对象包含一组由 SingleAnnotation 类定义的单个注释(图 1B),其中每个单独注释都包含一种特定类型的图形,该图形由 AnnotationFunction 类进一步定义。AnnotationFunction 类提供了一种灵活的方式来定义新的注释图形,更重要的是,自定义的注释图形可以根据主热图而进行自动重新排序和切分。
ComplexHeatmap 的主要特点是它支持水平或垂直连接一组热图和注释,以便于可视化不同数据源之间的关联。HeatmapList 类是一组热图和注释的容器(图 1C),它会自动调整多个热图和注释中行或列的对应关系。

图1. ComplexHeatmap 软件包的模块化设计
(A) 一个具有多个组件的单个热图。(B) 一列热图注释。(C) 一个热图列表。
单个热图
ComplexHeatmap 为配置单个热图提供了丰富的功能。构造函数 Heatmap() 用以生成单个热图,并且返回一个 Heatmap 类的对象。Heatmap() 中唯一的强制参数是一个矩阵,其可以是数字型或者字符型。Heatmap() 提供了大量用于自定义热图的附加参数。除了其他热图包中也提供的常见功能外,Heatmap() 还具有如下列出的这些独特功能。
⬤ 灵活地聚类和矩阵排序
在常规的数据分析过程中,热图通常伴随着对其矩阵的层次聚类,从而使得具有相似模式的特征被放置在邻近的位置,并且可以很容易地从热图上的颜色中识别出来。在 Heatmap() 中,可以通过多种方式指定层次聚类:1. 通过预定义的距离方法,例如“euclidean”或“pearson”,2. 通过一个距离函数,其可以通过两个向量来计算距离,或者直接从一个输入矩阵计算距离, 3. 通过将矩阵作为输入并返回一个dendrogram对象的聚类函数,4. 通过一个聚类对象,例如 hclust 对象或 dendrogram 对象。最后一种方法尤其有用,因为用户可以使用由其他软件包生成的或编辑的 dendrogram 对象。例如,使用 dendextend 包 [21]用不同的颜色渲染 dendrogram 的分支以突出显示子树,或者可以在 dendrogram 的节点上添加特定符号,然后渲染后的 dendrogram 对象可以直接在 Heatmap() 中使用(图2A )。
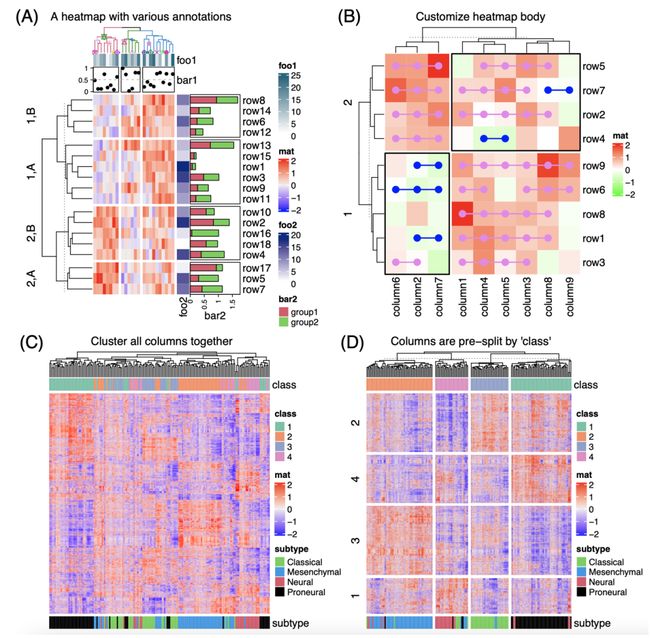
图2. 单个热图的使用
(A) 一个具有行和列注释的热图。热图的列被3个组的k均值聚类切分,行被一个分类变量和2个组的k均值聚类综合切分。(B) 对热图进行个性化修饰。图 A 和 B 中的数据是随机生成的。(C) 热图没有进行行和列的切分。(D) 热图进行行和列的切分。图 C 和 D 中使用的是同一个矩阵。
树状图通常表示为二叉树,其中两个分支的顺序在某个节点上的次序是任意的。旋转某个节点上的两个分支并不会改变其数学表示,但会影响 dendrogram 中元素的全局排序。因此,选择一个旋转 dendrogram 分支的合适的方法,或者换句话说,重新排序 dendrogram ,有助于在热图中将具有相似模式的矩阵行或列彼此靠近,以此提高可视化的效果。在默认情况下, Heatmap() 使用 reorder.dendrogram() 函数根据 dendrogram 分支的子矩阵的平均值对 dendrogram 重新排序,例如,在 dendrogram 的每个节点上,一个平均值较小的分支总是放在该节点的左侧。Heatmap() 也支持 dendrogram 对象,因此,其他 dendrogram 重新排序的方法可以很容易地集成其中。用户可以首先生成一个 dendrogram 对象,然后应用特定的 dendrogram 重新排序方法,例如来自 dendsort 包 [22],最后将它们集成到 Heatmap() 中。
注意层次聚类只是对热图的行和列进行重新排序的一种特殊方法。当然也可以使用计算矩阵的行和列顺序的其他方法。Heatmap() 允许用户设置数字索引或字符索引来重新排序热图。用于排序矩阵的流行软件包有 seriation [23] 和 biclust [24]。
⬤ 热图切分
热图切分是突出显示分组模式的一种有效方法。由于在层次聚类过程中,当向当前 dendrogram 中添加新的叶(leaf)或子树(sub-dendrogram)时,计算只是基于dendrogram中已经存在的元素,而不是矩阵中的所有所有元素。如果某些数据集中分组只有中等水平的差异,那么这会削弱它们的可视化的效果。热图切分可以极大地提高分组模式的可区分性。ComplexHeatmap 提供了多种方式将热图切分为行和列上的“切片”(slice)(图 2A-B):1. 设置 k均值聚类,其中支持重复运行 k均值聚类若干次以获得一个一致性k均值聚类结果,以减少随机性的影响;2. 设置一个包含预定义分组信息的分类变量。变量可以是一个向量或一个数据框,然后热图被分类变量中所有水平的组合切分;3. 如果在热图上已经应用了层次聚类,则可以指定单个数字,然后 cutree() 函数被用来来切割 dendrogram 。对于前两种拆分方法,如果启用了聚类,则首先在每个热图切片内执行层次聚类,然后根据它们的平均值对热图切片应用第二次聚类,用以显示切片级别的层次关系。
例如,图 2C 中的热图显示了在具有四个组别的胶质母细胞瘤数据集 [25] 中具有显着差异表达的基因(作为顶部注释)。这四个组别是通过一致性聚类(consensus clustering)预测的,分析表示分类的结果非常稳定 [26]。稳定的分类结果也得到了 t-SNE 分析的支持,其中四组别能够被很好地分开(图 S1)。但是,如果通过对所有样本直接应用层次聚类,那么四个组别并没有像预期的那样很好地分离,其中组别 3(蓝色)和 4(紫色)中的一些样本混合在一起。在图 2D 中,使用相同的矩阵,首先按照分类结果对热图进行拆分,然后在每个列切片中分别应用层次聚类。作为比较,它确实提高了分组模式的效果。此外,在图 2D 中,行还通过 k均值聚类进行切分。现在能够很容易的观察到组别特异性基因的表达模式。
⬤ 渲染热图为栅格图像
当我们制作所谓的“高质量图形”时,我们通常将图形保存为矢量图形,例如以格式 pdf 或 svg保存。矢量图形存储了每个图形元素的详细信息,因此,如果将由巨大矩阵生成的热图保存为矢量图形,那么文件将会非常大。完整的图像将需要很长时间才能被图像查看器渲染。由于图形设备的尺寸和分辨率有限,对于大型热图来说,热图中的相邻网格实际上会被合并为单个像素。因此,需要一种有效减少原始图像的方法,而不必保留巨大热图的所有细节。
栅格化是一种将图像转换为红-绿-蓝 (RGB) 值的颜色矩阵的方法。假设热图矩阵有 nr 行和 nc 列。当它在某个图形设备(例如屏幕设备)上绘制时,相应的热图主体分别使用 pr 和 pc 像素作为行和列。当 nr > pr 和/或 nc > pc 时,矩阵中的多个值被映射到单个像素上,其中 nr 和/或 nc 可以通过某种方法缩减为 pr 和/或 pc。ComplexHeatmap 提供了三种方法通过栅格化来缩减热图中的图形:1. 首先将热图写入一个分辨率为 pr × pc 的临时 png 图像,然后将临时图像读取为栅格对象并填充回热图。这种方法其实在 png 设备上进行了图像缩减。2. 首先将原始矩阵缩减为 pr × pc 的大小,那么缩减之后的矩阵中的单个值可以对应一个不同的像素。可以使用一些特定方法对矩阵进行缩减,例如从子矩阵中取平均值或随机值。3.首先生成一个分辨率为 nr × nc 的临时图像,然后使用 magick 包将图像缩小到 pr × pc 大小,最后将缩小后的图像作为栅格对象读取并填充到热图中。magick 包提供了大量调整图像大小的方法,并且ComplexHeatmap 都支持这些方法。在 ComplexHeatmap 一书的“Section 2.8 Heatmap as raster image”中,读者可以找到不同图像缩减方法的详细视觉比较。
⬤ 热图个性化
默认情况下,热图主体是由具有不同颜色的单元格组成。ComplexHeatmap 允许用户通过添加新的图层来个性化热图。Heatmap() 中的参数 cell_fun 和 layer_fun 可用于在绘制热图时将自定义的图形添加到热图单元格上(图 2A)。这两个参数的功能基本相同,但是 layer_fun 可以看做是 cell_fun 的矢量化版本。如果热图很大,使用 layer_fun 会使绘制速度更快。ComplexHeatmap 还提供了decorate_*() 系列函数,例如 decorate_annotation(),这些函数可以在热图绘制后将图形添加到任何热图组件中。在 ComplexHeatmap 中,每个热图组件都有自己的绘图区域,热图绘制结束后仍会记录它们。decorate_*() 可以返回到特定的绘图区域,然后在其中添加自定义图形。
正如后面部分将介绍的,3D 热图、oncoPrint 和 UpSet plot 都使用了 layer_fun 进行实现。而密度分布热图和富集热图则使用了 decorate_heatmap_body() 来部分增强其可视化效果。
⬤ 灵活地设置颜色和图例
热图中,颜色是主要用来映射到数据上的元素。ComplexHeatmap 允许通过设置一个颜色映射函数,指定断点和相应颜色来对矩阵中的值和颜色之间进行精确的映射。例如,用户可以定义一个对称于零的颜色映射函数,这有助于识别上调和下调基因的表达,或者用户可以为不同的热图定义相同的颜色映射函数,以使颜色在热图之间具有可比性。ComplexHeatmap 还允许对热图图例进行灵活的配置,例如多配色方案图例和具有自定义图形的图例。读者请参考 ComplexHeatmap 书中的“Chapter 5. Legends”以获得更多信息。
热图注释
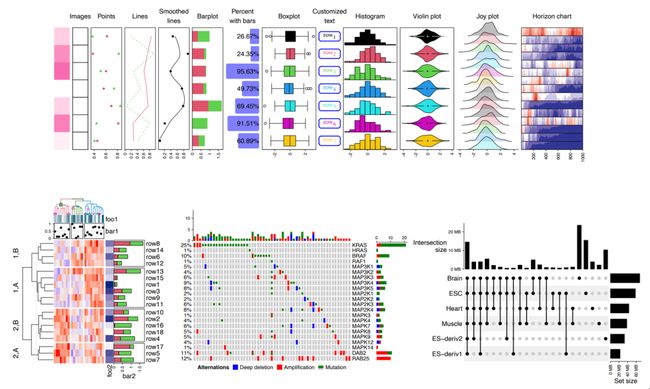
热图注释是热图的重要组成部分。它不仅显示与热图行和列相关的附加信息,而且还允许使用更多类型的图形进行可视化。ComplexHeatmap 为内置的热图注释图形以及新的自定义注释图形提供了灵活的支持。在图 3A 中,我们展示了 ComplexHeatmap 中部分默认支持的注释图形(从左到右):
1. 类似热图的注释。在 ComplexHeatmap 中它们被称为“简单注释”。它可以可视化数值型或者字符型的向量或者矩阵。
2. 图像注释。它支持多种格式的图像,例如 png、svg、pdf 或 jpg。
3. 散点注释。它支持单个数值向量或数值矩阵。
4. 线条注释。它支持单个数值向量或数值矩阵。
5. 平滑线注释。通过 loess 方法对一组或者多组散点进行平滑。
6. 柱状图注释。它也支持堆积柱状图。
7. 百分比注释。它同时包含文本和柱状图。
8. 箱线图注释。
9. 文字注释。它支持使用 gridtext 包构建自定义样式的文本。
10. 直方图注释。
11. 小提琴图注释。它用来可视化一组分布。分布也可以通过密度分布图或热图进行可视化。
12. Joy plot 注释。在其中,分布的峰值可以扩展到邻近的绘图区域中。
13. Horizon chart 注释 [27]。

图3. 各种热图注释
(A) ComplexHeatmap 中支持的一些热图注释图形。(B) 标记注释。(C) 连接注释。(D) 文本框注释。图 A 至图 D 中的数据都是随机生成的
所有内置的注释图形都由以 anno_ 前缀命名的函数实现,例如,用于散点注释的 anno_points()。除了上面列出的注释之外,ComplexHeatmap 还支持更复杂的注释。例如,anno_mark() 支持所谓的“标记注释”,它可以添加文本标记与部分行或者列对应(图 3B)。ComplexHeatmap 中的 anno_link() 支持所谓的“连接注释”,它可以将一组独立的绘图区域与热图中的行或者列对应。连接注释提供了一种通用的解决方案,可以将更多的自定义图形关联到热的行或列上。在图 3C 中,我们创建了三个 ggplot2 图形用以显示三个列组中值的分布,但仅在各组选定的行中。在图 3D 中,单词列表与每个行组相关联,其中字体大小对应于单词的重要性。这个文本框注释的功能可以通过函数 anno_textbox() 实现,并已被用于在 simplifyEnrichment 软件包 [28] 中用以总结一组基因的普遍的生物学功能。
构造函数 HeatmapAnnotation() 接受名称-值对的多个注释。简单注释可以直接设置为向量、矩阵或数据框。其他复杂的注释应通过函数 anno_*() 来指定。下面的例子中,我们展示了一个带有四个不同注释图形的热图注释。
ha = HeatmapAnnotation(
foo = runif(10),
bar = sample(letters[1:4], 10, replace = TRUE),
pt = anno_points(runif(10)),
txt = anno_text(month.name[1:10])
)热图的行注释应该使用一个额外的参数 which = "row" 或使用辅助函数 rowAnnotation() 来设置。ComplexHeatmap 提供了大量的注释图形,不过,ComplexHeatmap 还额外提供了创建自定义标注图形的接口。读者可以参阅 ComplexHeatmap 文档中的“Section 3.20 Implement new annotation functions”以获取更多详细信息。
热图列表
ComplexHeatmap 的一个重要的特性是它支持连接多个热图和热图注释,以便可视化多种信息之间的关联。ComplexHeatmap 提供了一种使用运算符 + 连接热图的简单语法。该表达式返回一个 HeatmapList 对象,直接执行 HeatmapList 对象会绘制热图。一个示例用法如下:
Heatmap(...) +
Heatmap(...) +
rowAnnotation(...)我们之前介绍了热图注释作为单个热图的组件。如上面的代码所示,行注释也可以独立连接到热图列表中。热图列表可以通过运算符 %v% 进行垂直连接。连接的热图和注释的数量可以是任意的。所有热图的排序和切分由主热图进行调整。主热图默认为第一个数字型的热图,或者用户可以自由指定。
Heatmap(...) %v%
Heatmap(...) %v%
HeatmapAnnotation(...)⬤ 可视化DNA甲基化和基因表达之间的关联
图 4A 展示了在一个随机生成但基于未发表研究中发现的模式的数据集上的复杂热图可视化。它可视化了 DNA 甲基化、基因表达、增强子和基因相关信息之间的复杂关联。在热图中,每一行对应于一个差异甲基化区域(DMR,它是一个在肿瘤和对照样本之间有显著甲基化差异的基因组区域)或与 DMR 对应的其他基因组属性。在图 4A 中从左到右有以下热图和注释:
1. DMR 中甲基化水平的热图。
2. 显示差异甲基化变化方向的单列热图。“Hyper”表示肿瘤样本中的甲基化程度较高,“hypo”表示肿瘤样本中的甲基化程度较低。
3. 基因表达的热图。这些基因是 DMR 最邻近的基因。
4. DMR 中的甲基化和对应基因的表达的 Pearson 相关性检验的 p 值的单列热图。
5. 基因类型的单列热图,例如蛋白质编码基因或 lincRNA?
6. DMR 在基因组中位置的单列热图,例如,在启动子或基因间区域?
7. DMR 到相关基因的 TSS 之间的距离,使用散点热图注释。
8. 增强子和 DMR 之间覆盖程度的热图。该值衡量每一个增强子被 DMR 覆盖的比例。
在图 4A 中,热图列表由差异甲基化方向和k均值聚类进行切分。k均值聚类分组是为了区分高甲基化组和低甲基化组。复杂热图显示了高度甲基化的 DMR 富集在基因间区域和基因内区域,但是很少与增强子重叠(行组“2,hypo”和“2,hyper”),而相比之下,低甲基化的 DMRs富集在启动子和增强子中( 行组“1,hypo”和“1,hyper”)。这可能意味着增强子和与低甲基化相关,并且增强子中的甲基化变化可能会影响它们对相关基因的转录活性变化。

图4. 复杂热图可视化
(A) 对 DNA 甲基化,基因表达和相关的基因组信息之间关联的可视化。为简单起见,图 A 中只包含了甲基化变化和基因表达负相关的 DMR。(B) 可视化多组学研究中各种描述统计量之间的关联。
⬤ 可视化多组学研究中多种描述统计量之间的关联
多组学研究整合来自基因组学、转录组学或表观基因组学的数据,以从不同层面寻找生物系统中的新关联。因此,正确有效地可视化这些不同数据类型之间的潜在联系是非常重要的。图 4B 展示了一种典型的对全局(landscape)统计量的可视化,其中基于单一数据类型或者多种数据类型的不同统计数据展示为一组热图和注释图形。图 4B 基于一个对胶质母细胞瘤的研究数据 [29],该研究利用 DNA 甲基化、基因表达和组蛋白修饰数据研究了四种亚型(图 4B 中索引为 1 至 4)之间的表观基因组差异。该研究生成了四组 DMR,其中每组 DMR 对一个亚型中的甲基化与正常样本进行比较。图 4B 使用热图和注释可视化 DMR 的各种基因组属性,另外按甲基化变化的方向对热图进行切分。图 4B 中从左到右有如下图形:
1. 肿瘤样本和正常样本中 DMR 的平均甲基化热图。
2. 每个类别中 DMR 数量,使用柱状图。
3. 与最邻近的基因表达显著相关的 DMR 的百分比,使用堆积柱状图。
4. DMR 到最近基因的转录起始位点 (TSS) 的距离,使用堆积柱状图。
5. 与基因或基因间区域重叠的 DMR 的百分比,使用堆积柱状图。
6. 与 CpG 岛 (CGI) 或 CGI shore重叠的 DMR 的百分比,使用堆积柱状图。
7. DMR 富集到基因组特征区域的热图。正值意味着过度富集(over-representation)。以 Jaccard 系数为统计量,通过随机重排DMR在基因组中的位置来获得随机条件下Jaccard 系数的分布。最终 z 值作为热图上的统计量,计算为(观测值 - 期望值)/ 标准差。
8. 与各种染色质状态重叠的 DMR 的百分比,使用堆积柱状图。
9. DMR 富集到染色质状态(chromatin states)的热图。同样,z 值作为热图上的统计量。
在图 4B 的热图列表中,可以直接观察到各组特定 DMR 的不同特征。例如,hyper-DMRs 在甲基化和基因表达之间具有更多的负相关性,hypo-DMRs 更多的位于基因间区域和非活性染色质状态。总而言之,这种可视化能够迅速揭示出复杂研究中的潜在关联。
高水平图形
ComplexHeatmap 的灵活性允许用户能够在具有类似矩阵结构的数据上实现新的高水平图形。ComplexHeatmap 已经实现了一些高级图形功能,我们将在以下小节中介绍。所有这些功能基本上都是热图的特定形式, 它们本质上是 Heatmap 对象,因此它们可以和到一般的热图和注释连接以形成复杂的可视化。
⬤ 密度热图
为了可视化矩阵或列表中的数据分布,我们通常使用箱线图或小提琴图。然而,当有大量的分布时,箱线图或小提琴图将不是有效的可视化方法。densityHeatmap() 函数使用颜色来映射分布的密度值,因此它能够可视化大量的分布(图 5A)。在 densityHeatmap() 中,分布之间的相似性可以用 Kolmogorov-Smirnov 距离来衡量。
⬤ 三维热图
通常不建议使用三维 (3D) 可视化 [30],但在特定场景中,3D可视化会非常有用。ComplexHeatmap 支持将普通热图转换为 3D 热图。3D 热图可以通过 Heatmap3D() 函数绘制,它接受与 Heatmap() 相同的参数集,只是2D的格子现在变成了3D的柱子(在二维空间的投影)。图 5A 中的密度分布也可以可视化为3D柱状图(图 5B)。我们建议在3D 热图可视化中,同时将颜色和柱子的高度都映射到数据上。
⬤ oncoPrint
oncoPrint() 函数可视化存在于一组基因和多个病人中的基因组异变事件,例如单碱基突变 (SNV)、片段插入或缺失 (Indels) 或拷贝数变异 (CNV)。oncoPrint() 提供了一种通用解决方案,其中用户可以自定义各种基因组异变所对应的图形(图 5C)。默认情况下,基因按在病人中异变的总数进行排序,而病人被重排序以显示基因在病人中的互斥性。由于 oncoPrint() 返回一个 Heatmap 对象,那么它可以直接和其他基因组数据集的热图进行连接,例如基因表达,以显示更复杂的基因组关联。
⬤ UpSet plot
与传统方法(即维恩图)相比,UpSet 图 [31] 提供了一种更有效的方法来可视化大量集合之间的关系。ComplexHeatmap 中的 UpSet() 函数提供了一个对UpSet plot原始工具 [32] 的增强实现。此外,UpSet() 能够处理来多个基因组区间列表,这有助于揭示例如组织特异性染色质修饰的模式(图 5D)。
⬤ 基因组水平图形
基因组水平的热图经常用于基因组学研究,例如,用于可视化全局拷贝数变异概况 [33]。制作基因组级别的热图的关键是对基因组进行区间化,并将各种基因组信号归一化到基因组区间中以形成矩阵,然后就可以对其进行正常的热图可视化。在图 5E 中,我们展示了一个基因组水平的可视化,其中包含两个热图和多个作为热图注释的图形。

图5. ComplexHeatmap 中支持的高水平图形
(A) 分布密度热图。前10列的随机数据来自于正态分布,后10列的随机数据来自于均匀分布。(B) 3D 频度热图。其中使用的数据来自于图 A。(C) oncoPrint。使用了来自于cBioPortal的肺腺癌数据。由于图片的大小限制,我们只展示了部分基因和部分病人。(D) UpSet plot。六个人类组织的H3K4me3 ChIP-seq 的数据都来自于 Roadmap 项目。(E) 基因组水平图形。数据是随机生成的
与其他软件包的集成
⬤ Enriched heatmap
富集热图(Enriched heatmap)专门可视化特定类型的基因组信号在某种基因组特征区间上的富集 [34]。例如,研究染色质修饰如何在基因 TSS 周围富集,或者 DNA 甲基化如何在 CGI 周围显示低甲基化。EnrichedHeatmap 包 [6] 建立在 ComplexHeatmap 之上,它为显示两种类型的基因组特征的空间关系提供了通用解决方案。它还实现了一个特殊的热图注释函数 anno_enriched(),它能够绘制所有基因组特征的平均富集度。与其他类似工具相比,EnrichedHeatmap的独特之处在于,它能够处理离散的基因组信号,例如全基因组根据染色质状态的分隔(chromatin segmentation)。更重要的是,富集热图也是一个Heatmap对象,因此它支持 Heatmap 类的所有特征,例如热图切分和与更多热图连接。
图 6 展示了染色质状态分布,基因 TSS 周围的 DNA 甲基化分布,以及相关基因表达的复杂可视化。数据来自于 Roadmap 项目 [35]。热图按照行被分为三组,其中的 TSS 分别处于 active 状态、bivalent 状态和 inactive 状态。通过复杂热图,可以很容易地观察到 active TSS 与低甲基化相关,并且相应的基因高表达。Bivalent TSS,虽然也是低甲基化的,但基因的表达较低。作为比较,inactive TSS 几乎完全甲基化,但是相应基因的表达通常被沉默。
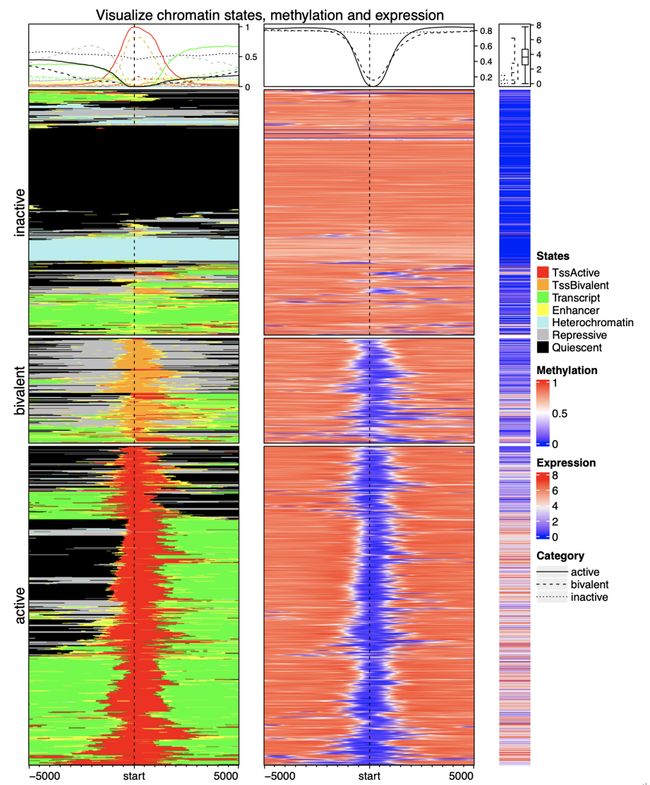
图6. 一组富集热图和常规热图
从左至右依次是染色质状态的热图,DNA 甲基化的热图和基因表达的热图。数据来自于 Roadmap 项目
⬤ InteractiveComplexHeatmap
ComplexHeatmap 只生成静态图形。R包 InteractiveComplexHeatmap [36] 可以将静态热图非常容易地转换为交互式的 Shiny 应用程序,在其中,用户可以直接通过点击或者区域选择与热图进行交互。从静态热图到交互式热图的转换可以通过 htShiny() 函数完成,在静态热图生成后,htShiny() 可以直接不带任何参数执行。此功能适用于所有由 ComplexHeatmap 生成的热图。此外,InteractiveComplexHeatmap 支持灵活的设计交互式热图的用户界面以及用户在热图上的操作响应。
总 结
复杂热图是一种用来揭示多种信息之间复杂关联的强大的可视化方法。在本篇论文中,我们系统性地展示了 ComplexHeatmap 软件包的各种功能。我们相信,ComplexHeatmap 会继续成为生物信息学乃至数据科学领域中的一个强有用的工具,用来有效的展示隐藏在海量数据背后的结构。
致谢
本项研究受到National Center for Tumor Diseases (NCT) Molecular Precision Oncology Program(德国)支持。
利益冲突
作者没有申明任何利益冲突。
作者贡献
顾祖光:研究课题的提出和设计, 软件编写,可视化,数据分析,论文编写,修订和审阅。
代码和数据可用性
ComplexHeatmap 的稳定版本发布在 https://bioconductor.org/packages/ComplexHeatmap/
开发者版本发布在https://github.com/jokergoo/ComplexHeatmap,
文档发布在
https://jokergoo.github.io/ComplexHeatmap-reference/book/
论文中绘制图1到图6的代码发布在
https://github.com/jokergoo/ComplexHeatmap_v2_paper_code
ORCID
0000-0002-7395-8709 (Zuguang Gu)
作者简介

顾祖光(第一兼通讯作者)
● 德国癌症研究中心staff scientist
●在生物信息学相关期刊,例如Bioinformatics, Nucleic Acids Research, Briefings in Bioinformatics,Genomics Proteomics & Bioinformatics等,和其他高影响力期刊,例如Nature Immunology, Nature Communications, Cancer Discovery,发表论文30余篇,累计引用7千余次。独立开发众多生物软件包,总下载量超3百万次。
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:[email protected]
微信公众号
iMeta
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























机器学习
后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集


