7. 生信技能树——TCGA癌症数据2
一. “生存分析前的数据整理”
1.读入数据
表达矩阵只需要tumor数据,不要normal,将其去掉,新表达矩阵数据命名为exprSet;
临床信息需要进一步整理,成为生存分析需要的格式,新临床信息数据命名为meta。
由于不同癌症的临床信息表格列名可能不同,这里的代码需要根据实际情况修改。
rm(list=ls())
proj = "TCGA-KIRC"
load(paste0(proj,".Rdata"))
library(stringr)
2.整理表达矩阵
不需要正常样本;使用logCPM或logTPM数据
exprSet=log2(edgeR::cpm(exp[,Group=='tumor'])+1) ## 可以仿照这个将RNA_seq测序的count数据转换成cpm数据,即表达矩阵,这个矩阵可用来画热图
ncol(exprSet)
因前面的差异分析过滤标准有宽有严,保险起见,这里可以再次进行基因过滤,至少要在50%的样本里表达量大于0。
k = apply(exprSet,1, function(x){sum(x>0)>0.5*ncol(exprSet)});table(k) # 对行进行计算,每行中至少有一半的样本的值大于0
exprSet = exprSet[k,]
nrow(exprSet)
3.整理生存信息和临床信息
xena将生存信息和临床信息分开了。后续构建模型需要纳入一些临床信息,所以要合并到一起。
library(dplyr)
meta = left_join(surv,clinical,by = c("sample"= "submitter_id.samples"))
# 去掉表达矩阵里没有的样本
library(stringr)
k = meta$sample %in% colnames(exprSet);table(k)
meta = meta[k,]
# 去掉生存信息不全或者生存时间小于30天的样本,样本纳排标准不唯一,且差别很大
k1 = meta$OS.time >= 30;table(k1)
k2 = !(is.na(meta$OS.time)|is.na(meta$OS));table(k2)
meta = meta[k1&k2,]
# 选择有用的列
tmp = data.frame(colnames(meta))
meta = meta[,c(
'sample',
'OS',
'OS.time',
'race.demographic',
'age_at_initial_pathologic_diagnosis',
'gender.demographic' ,
'tumor_stage.diagnoses'
)]
dim(meta)
rownames(meta) <- meta$sample
meta[1:4,1:4]
#简化meta的列名
colnames(meta)=c('ID','event','time','race','age','gender','stage')
#空着的值、not reported改为NA
meta[meta==""|meta=="not reported"]=NA
3.实现表达矩阵与临床信息的匹配
有的病人会有两个或两个以上的肿瘤样本,就有重复。两种可行的办法:
(1)以病人为中心,对表达矩阵的列按照病人ID去重复,每个病人只保留一个样本。
exprSet = exprSet[,sort(colnames(exprSet))]
k = !duplicated(str_sub(colnames(exprSet),1,12));table(k)
exprSet = exprSet[,k]

(2)以样本为中心,如果每个病人有多个样本则全部保留。(删掉上面这一段代码即可)
#调整meta行名与exprSet列名一一对应
s = intersect(rownames(meta),colnames(exprSet))
exprSet = exprSet[,s]
meta = meta[s,]
identical(rownames(meta),colnames(exprSet))
4. 整理生存分析的输入数据
生存分析的输入数据里,要求结局事件必须用0和1表示,0表示活着,1表示死了;
生存时间的单位(月);
table(meta$event)
range(meta$time)
meta$time = meta$time/30
range(meta$time)
抹除stage里的重复信息
head(meta$stage)
meta$stage = meta$stage %>%
str_remove("stage ") %>%
str_to_upper()
table(meta$stage,useNA = "always")
# 不需要ABC可以去掉,需要的话就保留,不运行下面这句
meta$stage = str_remove(meta$stage,"A|B|C")
head(meta)
save(meta,exprSet,proj,file = paste0(proj,"_sur_model.Rdata"))
二.生存分析
1.准备输入数据
rm(list = ls())
proj = "TCGA-KIRC"
load(paste0(proj,"_sur_model.Rdata"))
ls()
exprSet[1:4,1:4]
meta[1:4,1:4]
2.KM-plot
简单版本和进阶版本
library(survival)
library(survminer)
sfit <- survfit(Surv(time, event)~gender, data=meta)
ggsurvplot(sfit,pval=TRUE)
ggsurvplot(sfit,
palette = "jco",
risk.table =TRUE,
pval =TRUE,
conf.int =TRUE)


连续型信息怎么作KM分析?例如年龄,基因?
连续型数据的离散化
年龄
group = ifelse(meta$age>median(meta$age,na.rm = T),"older","younger")
table(group)
sfit=survfit(Surv(time, event)~group, data=meta)
ggsurvplot(sfit,pval =TRUE, data = meta, risk.table = TRUE)

基因
g = rownames(exprSet)[1];g
meta$gene = ifelse(exprSet[g,]> median(exprSet[g,]),'high','low')
sfit=survfit(Surv(time, event)~gene, data=meta)
ggsurvplot(sfit,pval =TRUE, data = meta, risk.table = TRUE)

3.log-rank test
KM的p值是log-rank test得出的,可以批量操作
logrankfile = paste0(proj,"_log_rank_p.Rdata")
if(!file.exists(logrankfile)){
log_rank_p <- apply(exprSet , 1 , function(gene){
meta$group=ifelse(gene>median(gene),'high','low')
data.survdiff=survdiff(Surv(time, event)~group,data=meta)
p.val = 1 - pchisq(data.survdiff$chisq, length(data.survdiff$n) - 1)
return(p.val)
})
log_rank_p=sort(log_rank_p)
save(log_rank_p,file = logrankfile)
}
load(logrankfile)
table(log_rank_p<0.01)
table(log_rank_p<0.05)
4.批量单因素cox
coxfile = paste0(proj,"_cox.Rdata")
if(!file.exists(coxfile)){
cox_results <-apply(exprSet , 1 , function(gene){
meta$gene = gene
#可直接使用连续型变量
m = coxph(Surv(time, event) ~ gene, data = meta)
#也可使用二分类变量
#meta$group=ifelse(gene>median(gene),'high','low')
#meta$group = factor(meta$group,levels = c("low","high"))
#m=coxph(Surv(time, event) ~ group, data = meta)
beta <- coef(m)
se <- sqrt(diag(vcov(m)))
HR <- exp(beta)
HRse <- HR * se
#summary(m)
tmp <- round(cbind(coef = beta,
se = se, z = beta/se,
p = 1 - pchisq((beta/se)^2, 1),
HR = HR, HRse = HRse,
HRz = (HR - 1) / HRse,
HRp = 1 - pchisq(((HR - 1)/HRse)^2, 1),
HRCILL = exp(beta - qnorm(.975, 0, 1) * se),
HRCIUL = exp(beta + qnorm(.975, 0, 1) * se)), 3)
return(tmp['gene',])
#return(tmp['grouphigh',])#二分类变量
})
cox_results=as.data.frame(t(cox_results))
save(cox_results,file = coxfile)
}
load(coxfile)
table(cox_results$p<0.01)
table(cox_results$p<0.05)
lr = names(log_rank_p)[log_rank_p<0.01];length(lr)
cox = rownames(cox_results)[cox_results$p<0.01];length(cox)
length(intersect(lr,cox))
save(lr,cox,file = paste0(proj,"_logrank_cox_gene.Rdata"))
5.lasso回归
1.准备输入数据
rm(list = ls())
proj = "TCGA-KIRC"
load(paste0(proj,"_sur_model.Rdata"))
ls()
exprSet[1:4,1:4]
meta[1:4,1:4]
load(paste0(proj,"_logrank_cox_gene.Rdata"))
exprSet = exprSet[cox,]
2.构建lasso回归模型
输入数据是表达矩阵(仅含tumor样本)和每个病人对应的生死(顺序必须一致)。
x=t(exprSet) # x行名为样本,列名为基因
y=meta$event
library(glmnet)
2.1挑选合适的λ值
- Lambda 是构建模型的重要参数。他的大小关系着模型选择的基因个数
#调优参数
set.seed(1006) # 选取不同的数,画出来的效果不同
cv_fit <- cv.glmnet(x=x, y=y)
plot(cv_fit)
#系数图
fit <- glmnet(x=x, y=y)
plot(fit,xvar = "lambda")
两条虚线分别指示了两个特殊的λ值,一个是lambda.min,一个是lambda.1se,这两个值之间的lambda都认为是合适的。lambda.1se构建的模型最简单,即使用的基因数量少,而lambda.min则准确率更高一点,使用的基因数量更多一点。


2.2 用这两个λ值重新建模
model_lasso_min <- glmnet(x=x, y=y,lambda=cv_fit$lambda.min)
model_lasso_1se <- glmnet(x=x, y=y,lambda=cv_fit$lambda.1se)
选中的基因与系数存放于模型的子集beta中,用到的基因有一个s0值,没用的基因只记录了“.”,所以可以用下面代码挑出用到的基因。
head(model_lasso_min$beta,20)
choose_gene_min=rownames(model_lasso_min$beta)[as.numeric(model_lasso_min$beta)!=0]
choose_gene_1se=rownames(model_lasso_1se$beta)[as.numeric(model_lasso_1se$beta)!=0]
length(choose_gene_min)
length(choose_gene_1se)
save(choose_gene_min,file = paste0(proj,"_lasso_choose_gene_min.Rdata"))
save(choose_gene_1se,file = paste0(proj,"_lasso_choose_gene_1se.Rdata"))
3.模型预测和评估
newx参数是预测对象。输出结果lasso.prob是一个矩阵,第一列是min的预测结果,第二列是1se的预测结果,预测结果是概率,或者说百分比,不是绝对的0和1。
将每个样本的生死和预测结果放在一起,直接cbind即可。
lasso.prob <- predict(cv_fit, newx=x , s=c(cv_fit$lambda.min,cv_fit$lambda.1se) )
re=cbind(y ,lasso.prob)
head(re)
re=as.data.frame(re)
colnames(re)=c('event','prob_min','prob_1se')
re$event=as.factor(re$event)
ROC曲线
library(pROC)
library(ggplot2)
m <- roc(meta$event, re$prob_min)
g <- ggroc(m,legacy.axes = T,size = 1,color = "#2fa1dd")
auc(m) # Area under the curve: 0.9953
g + theme_minimal() +
geom_segment(aes(x = 0, xend = 1, y = 0, yend = 1),
colour = "grey", linetype = "dashed")+
annotate("text",x = .75, y = .25,
label = paste("AUC of min = ",format(round(as.numeric(auc(m)),2),nsmall = 2)),color = "#2fa1dd")
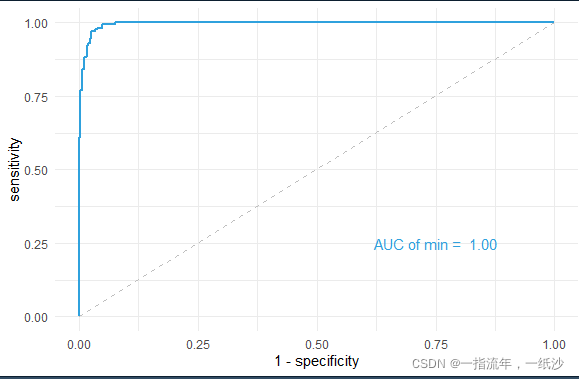
计算AUC取值范围在0.5-1之间,越接近于1越好。可以根据预测结果绘制ROC曲线。
两个模型的曲线画在一起
m2 <- roc(meta$event, re$prob_1se)
auc(m2) # Area under the curve: 0.9136
g <- ggroc(list(min = m,se = m2),legacy.axes = T,size = 1)
g + theme_minimal() +
scale_color_manual(values = c("#2fa1dd", "#f87669"))+
geom_segment(aes(x = 0, xend = 1, y = 0, yend = 1),
colour = "grey", linetype = "dashed")+
annotate("text",x = .75, y = .25,
label = paste("AUC of min = ",format(round(as.numeric(auc(m)),2),nsmall = 2)),color = "#2fa1dd")+
annotate("text",x = .75, y = .15,
label = paste("AUC of 1se = ",format(round(as.numeric(auc(m2)),2),nsmall = 2)),color = "#f87669")

5.切割数据构建模型并预测
5.1 切割数据
用R包caret切割数据,生成的结果是一组代表列数的数字,用这些数字来给表达矩阵和meta取子集即可。
library(caret)
set.seed(12345679)
sam<- createDataPartition(meta$event, p = .5,list = FALSE)
head(sam)
可查看两组一些临床参数切割比例
train <- exprSet[,sam]
test <- exprSet[,-sam]
train_meta <- meta[sam,]
test_meta <- meta[-sam,]
prop.table(table(train_meta$stage))
prop.table(table(test_meta$stage))
prop.table(table(test_meta$race))
prop.table(table(train_meta$race))

5.2 切割后的train数据集建模
和上面的建模方法一样。
#计算lambda
x = t(train)
y = train_meta$event
cv_fit <- cv.glmnet(x=x, y=y)
plot(cv_fit)

#构建模型
model_lasso_min <- glmnet(x=x, y=y,lambda=cv_fit$lambda.min)
model_lasso_1se <- glmnet(x=x, y=y,lambda=cv_fit$lambda.1se)
#挑出基因
head(model_lasso_min$beta)
choose_gene_min=rownames(model_lasso_min$beta)[as.numeric(model_lasso_min$beta)!=0]
choose_gene_1se=rownames(model_lasso_1se$beta)[as.numeric(model_lasso_1se$beta)!=0]
length(choose_gene_min)
length(choose_gene_1se)

4.模型预测
用训练集构建模型,预测测试集的生死,注意newx参数变了。
lasso.prob <- predict(cv_fit, newx=t(test), s=c(cv_fit$lambda.min,cv_fit$lambda.1se) )
re=cbind(event = test_meta$event ,lasso.prob)
re=as.data.frame(re)
colnames(re)=c('event','prob_min','prob_1se')
re$event=as.factor(re$event)
head(re)
再画ROC曲线
library(pROC)
library(ggplot2)
m <- roc(test_meta$event, re$prob_min)
g <- ggroc(m,legacy.axes = T,size = 1,color = "#2fa1dd")
auc(m) #Area under the curve: 0.7752
g + theme_minimal() +
geom_segment(aes(x = 0, xend = 1, y = 0, yend = 1),
colour = "grey", linetype = "dashed")+
annotate("text",x = .75, y = .25,
label = paste("AUC of min = ",format(round(as.numeric(auc(m)),2),nsmall = 2)),color = "#2fa1dd")
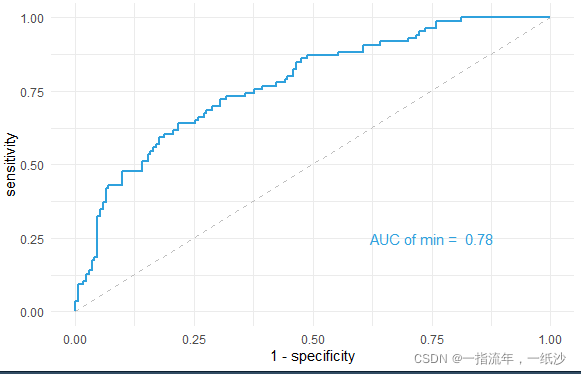
计算AUC取值范围在0.5-1之间,越接近于1越好。可以根据预测结果绘制ROC曲线。
两个模型的曲线画在一起
m2 <- roc(test_meta$event, re$prob_1se)
auc(m2) # Area under the curve: 0.7426
g <- ggroc(list(min = m,se = m2),legacy.axes = T,size = 1)
g + theme_minimal() +
scale_color_manual(values = c("#2fa1dd", "#f87669"))+
geom_segment(aes(x = 0, xend = 1, y = 0, yend = 1),
colour = "grey", linetype = "dashed")+
annotate("text",x = .75, y = .25,
label = paste("AUC of min = ",format(round(as.numeric(auc(m)),2),nsmall = 2)),color = "#2fa1dd")+
annotate("text",x = .75, y = .15,
label = paste("AUC of 1se = ",format(round(as.numeric(auc(m2)),2),nsmall = 2)),color = "#f87669")

6.cox-forest
1.准备输入数据
rm(list = ls())
proj = "TCGA-KIRC"
if(!require(My.stepwise))install.packages("My.stepwise")
load(paste0(proj,"_sur_model.Rdata"))
load(paste0(proj,"_lasso_choose_gene_1se.Rdata"))
g = choose_gene_1se
2.构建coxph模型
将用于建模的基因(例如lasso回归选中的基因)从表达矩阵中取出来,,可作为列添加在meta表噶的后面,组成的数据框赋值给dat。
library(stringr)
e=t(exprSet[g,])
colnames(e)= str_replace_all(colnames(e),"-","_")
dat=cbind(meta,e)
dat$gender=as.numeric(factor(dat$gender))
dat$stage=as.numeric(factor(dat$stage))
colnames(dat)
逐步回归法构建最优模型
输出结果行数太多,所以我注释掉了
library(survival)
library(survminer)
# 不能允许缺失值
dat2 = na.omit(dat)
library(My.stepwise)
vl <- colnames(dat)[c(5:ncol(dat))]
# My.stepwise.coxph(Time = "time",
# Status = "event",
# variable.list = vl,
# data = dat2)
使用输出结果里的最后一个模型
model = coxph(formula = Surv(time, event) ~ stage + age + AL357140.2 +
C1DP1 + HCCAT5 + AC131097.2 + LINC01522 + AC011497.2 + PROX1 +
AC021171.1 + INAFM2 + GREB1L + CCL22 + SLAMF9 + LINC01675 +
AP001893.3 + AC092296.1 + ZNF320 + MZT1P2 + CDC42BPG + AL157832.1 +
AC040934.1 + AC018659.8 + CHI3L2, data = dat2)
3.模型可视化-森林图
ggforest(model,data = dat2)
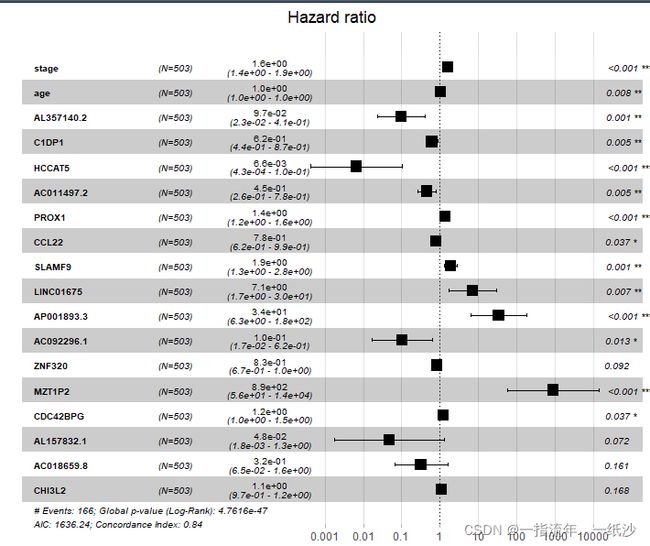
4.模型预测
fp <- predict(model,newdata = dat2)
library(Hmisc)
options(scipen=200)
with(dat2,rcorr.cens(fp,Surv(time, event)))
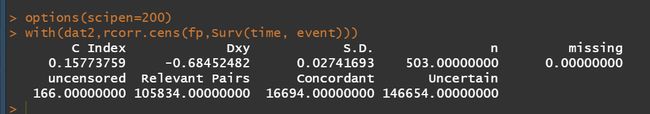
C-index用于计算生存分析中的COX模型预测值与真实之间的区分度(discrimination),也称为Harrell’s concordanceindex。C-index在0.5-1之间。0.5为完全不一致,说明该模型没有预测作用,1为完全一致,说明该模型预测结果与实际完全一致。
5.切割数据构建模型并预测
5.1 切割数据
用R包caret切割数据,生成的结果是一组代表列数的数字,用这些数字来给表达矩阵和meta取子集即可。
library(caret)
set.seed(12345679)
sam<- createDataPartition(meta$event, p = .5,list = FALSE)
train <- exprSet[,sam]
test <- exprSet[,-sam]
train_meta <- meta[sam,]
test_meta <- meta[-sam,]
5.2 切割后的train数据集建模
和上面的建模方法一样。
e=t(train[g,])
colnames(e)= str_replace_all(colnames(e),"-","_")
dat=cbind(train_meta,e)
dat$gender=as.numeric(factor(dat$gender))
dat$stage=as.numeric(factor(dat$stage))
colnames(dat)
library(My.stepwise)
dat2 = na.omit(dat)
vl <- colnames(dat2)[c(5:ncol(dat2))]
# My.stepwise.coxph(Time = "time",
# Status = "event",
# variable.list = vl,
# data = dat2)
model = coxph(formula = Surv(time, event) ~ stage + AC092651.1 + MZT1P2 +
NOC2LP2 + CCL22 + AC021171.1 + INAFM2 + LINC01522 + AC018630.2 +
STK19B + ZNF320 + GREB1L + NARF + SEMA3A + COL18A1_AS1 +
HCCAT5 + C1DP1 + AF230666.2 + LRFN1 + TGM3 + AC092296.1 +
CDC42BPG + RHNO1 + AC107982.3 + AL157832.1 + AC002070.1,
data = dat2)
5.3 模型可视化
ggforest(model, data =dat2)

5.4 用切割后的数据test数据集验证模型
e=t(test[g,])
colnames(e)= str_replace_all(colnames(e),"-","_")
test_dat=cbind(test_meta,e)
test_dat$gender=as.numeric(factor(test_dat$gender))
test_dat$stage=as.numeric(factor(test_dat$stage))
fp <- predict(model,newdata = test_dat)
library(Hmisc)
with(test_dat,rcorr.cens(fp,Surv(time, event)))
