GEO数据库挖掘--芯片数据--使用limma包
目录
- GEO数据库说明
- 下载GEO数据并加载表达矩阵
- ID转换
- 获取分组信息
- 表达矩阵质控
- 差异分析
- 结果可视化
- 富集分析
主要参考: https://www.jianshu.com/p/8dd7dc1e1719
一点点说明:
用基因芯片的手段来研究基因表达量的技术已经逐步被RNA-seq技术取代,但毕竟经历了十多年的发展了,在GEO或arrayexpress数据库里面存储的全球研究者数据都已经超过了50PB了!里面还是有非常多等待挖掘的地方!对基因表达芯片数据进行分析的主角就是limma,虽然一直都有各种差异分析统计方法提出来,但是limma包绝对是其中的佼佼者。
整体流程:

GEO数据库说明
文献中如果使用了GEO芯片,就一定会明确说明其使用的GSE编号。直接去文献里面找就可以了。
有三个概念需要理解
- GEO Platform (GPL):做芯片使用的平台。
- GEO Sample (GSM):样本信息
- GEO Series (GSE):数据
此处使用的数据为:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE42872
下载GEO数据并加载表达矩阵
一共三种方式:
- 直接下载rawdata ---- 基本没人这么干
- 下载Series Matrix File(s),再导入R
- 使用GEOquery包下载 ---- 推荐使用该方式
第一种方式不再说明。
第二种方式:
# 下载并解压Series Matrix File(s)
wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE42nnn/GSE42872/matrix/GSE42872_series_matrix.txt.gz
gzip -k -d GSE42872_series_matrix.txt.gz # -k:保留原来的gz文件
# 读取txt文件
a <- read.table(file = '1. Series_data/GSE42872_series_matrix.txt',
header = T, sep = '\t', quote = '', fill = T,
comment.char = '!')
# 把行号设置成第一列的值
rownames(a) <- a[,1]
# 去除ID_REF列,因为它与数据的列名是重复的。
a <- a[,-1] # 读取完成
第三种方式:
library(GEOquery)
# getGEO得到的是一个list,里面包含了很多信息,但只有第一个信息是芯片数据。
eSet <- getGEO('GSE42872',
destdir = '1. getGEO_data/', # 存放位置
getGPL = F) # 不需要平台信息,这个太考验网速
# 使用exprs(Retrieve expression data from eSets.)函数提取表达数据
exp <- exprs(eSet[[1]])
两种方法得到的表达矩阵是相同的(即:a == exp)
ID转换
官网下载是金标准,但官网不一定能打开,且只能一个一个下载;
NCBI可以下载,但文件太大。
推荐bioconductor包直接转换,常见的平台如下:
参考:http://www.bio-info-trainee.com/1399.html
gpl organism bioc_package
1 GPL32 Mus musculus mgu74a
2 GPL33 Mus musculus mgu74b
3 GPL34 Mus musculus mgu74c
6 GPL74 Homo sapiens hcg110
7 GPL75 Mus musculus mu11ksuba
8 GPL76 Mus musculus mu11ksubb
9 GPL77 Mus musculus mu19ksuba
10 GPL78 Mus musculus mu19ksubb
11 GPL79 Mus musculus mu19ksubc
12 GPL80 Homo sapiens hu6800
13 GPL81 Mus musculus mgu74av2
14 GPL82 Mus musculus mgu74bv2
15 GPL83 Mus musculus mgu74cv2
16 GPL85 Rattus norvegicus rgu34a
17 GPL86 Rattus norvegicus rgu34b
18 GPL87 Rattus norvegicus rgu34c
19 GPL88 Rattus norvegicus rnu34
20 GPL89 Rattus norvegicus rtu34
22 GPL91 Homo sapiens hgu95av2
23 GPL92 Homo sapiens hgu95b
24 GPL93 Homo sapiens hgu95c
25 GPL94 Homo sapiens hgu95d
26 GPL95 Homo sapiens hgu95e
27 GPL96 Homo sapiens hgu133a
28 GPL97 Homo sapiens hgu133b
29 GPL98 Homo sapiens hu35ksuba
30 GPL99 Homo sapiens hu35ksubb
31 GPL100 Homo sapiens hu35ksubc
32 GPL101 Homo sapiens hu35ksubd
36 GPL201 Homo sapiens hgfocus
37 GPL339 Mus musculus moe430a
38 GPL340 Mus musculus mouse4302
39 GPL341 Rattus norvegicus rae230a
40 GPL342 Rattus norvegicus rae230b
41 GPL570 Homo sapiens hgu133plus2
42 GPL571 Homo sapiens hgu133a2
43 GPL886 Homo sapiens hgug4111a
44 GPL887 Homo sapiens hgug4110b
45 GPL1261 Mus musculus mouse430a2
49 GPL1352 Homo sapiens u133x3p
50 GPL1355 Rattus norvegicus rat2302
51 GPL1708 Homo sapiens hgug4112a
54 GPL2891 Homo sapiens h20kcod
55 GPL2898 Rattus norvegicus adme16cod
60 GPL3921 Homo sapiens hthgu133a
63 GPL4191 Homo sapiens h10kcod
64 GPL5689 Homo sapiens hgug4100a
65 GPL6097 Homo sapiens illuminaHumanv1
66 GPL6102 Homo sapiens illuminaHumanv2
67 GPL6244 Homo sapiens hugene10sttranscriptcluster
68 GPL6947 Homo sapiens illuminaHumanv3
69 GPL8300 Homo sapiens hgu95av2
70 GPL8490 Homo sapiens IlluminaHumanMethylation27k
71 GPL10558 Homo sapiens illuminaHumanv4
72 GPL11532 Homo sapiens hugene11sttranscriptcluster
73 GPL13497 Homo sapiens HsAgilentDesign026652
74 GPL13534 Homo sapiens IlluminaHumanMethylation450k
75 GPL13667 Homo sapiens hgu219
76 GPL15380 Homo sapiens GGHumanMethCancerPanelv1
77 GPL15396 Homo sapiens hthgu133b
78 GPL17897 Homo sapiens hthgu133a
本文使用的是GPL6244,所以使用hugene10sttranscriptcluster即可。但是,前提是先把上面所有的包先安装好。如果不想全部安装,可以只安装自己需要的。
# 把上面的文件做成一个csv文件。运行下面的代码安装所有的包。
gpl_info=read.csv("GPL_info.csv",stringsAsFactors = F)
### first download all of the annotation packages from bioconductor
for (i in 1:nrow(gpl_info)){
print(i)
platform=gpl_info[i,4]
platform=gsub('^ ',"",platform) ##主要是因为我处理包的字符串前面有空格
#platformDB='hgu95av2.db'
platformDB=paste(platform,".db",sep="")
if( platformDB %in% rownames(installed.packages()) == FALSE) {
BiocManager::install(platformDB)
}
}
芯片对于每个基因都会设计多个探针,而我们下载的表达矩阵是submmiter处理之后的,其已经过滤掉很多表达较差的探针。当我们处理他人的数据时,默认是要认为submmiter处理的是对的。否则你就要下载作者的原始数据自己分析。
# 加载自己需要的包,注意一定要在最后添加.db。
library(hugene10sttranscriptcluster.db)
# 查看一下这个包的所有内容
ls("package:hugene10sttranscriptcluster.db")
# 提取probe_id和symbol
ids=toTable(hugene10sttranscriptclusterSYMBOL)
# 查看一下,作者保留的数据是不是每个id在芯片中都有对应的symbol
table(rownames(exp) %in% ids$probe_id)
# 过滤掉无法对应的probe_id
exp <- exp[rownames(exp) %in% ids$probe_id,]
# 重新排序一下ids中的探针顺序,使其与表达矩阵的顺序一致
ids=ids[match(rownames(exp),ids$probe_id),]
现在虽然已经有了exp和ids了,但是,由于一个基因有多个探针,所以现在的exp中还是会出现一个基因对应多个探针的情况。
# 查看一下,是不是一个基因只对应一个探针
table(sort(table(ids$symbol)))
# 输出结果为:
# 1 2 3 4 5 6 7 8 10 26
# 18094 596 130 15 7 4 1 2 1 1
从结果可以看出,还是有很多基因对应多个探针。那么对于这些有多个探针的基因,我们可以选择把所有探针的平均值作为其表达量。
# 通过ids中的symbol对数据进行分组,然后再从中选择最大的平均值
tmp = by(exp,
ids$symbol, # 通过ids中的symbol对数据进行分组
function(x) rownames(x)[which.max(rowMeans(x))]) # 选择最大的平均值作为基因的表达量。
probes = as.character(tmp)
exp = exp[rownames(exp) %in% probes,] # 过滤有多个探针的基因
现在的数据才可以算是一个tidy的数据,此时就可以将表达矩阵的探针id换成基因symbol了:
rownames(exp)=ids[match(rownames(exp),ids$probe_id),2]
获取分组信息
有两种方法:
- 使用pData (Retrieve information on experimental phenotypes recorded in eSet and ExpressionSet-derived classes.)获取每个样本的描述信息,并从中然后再使用stringr提取。
- 自己手动生成一个文件
第一种方法:
pd <- pData(eSet[[1]])
library(stringr)
group_list = ifelse(str_detect(pd$title,"Control")==TRUE,"contorl","treat")
group_list
第二种方法:
由于已经知道只有6个样品,前3个是control后3个是treat,直接:
group_list=c(rep("control",times=3),rep("treat",times=3))
group_list
表达矩阵质控
大概进行一下下面三个分析即可:
- 各个样本表达量分析
- 管家基因表达量分析
- PCA图-hclust图分析样本分组信息
- 各个样本表达量分析
首先分析一下每个样品平均的表达水平:
boxplot(exp)
 可以看到,每个样品的平均表达都在8左右。
可以看到,每个样品的平均表达都在8左右。
同时,也可以看出,所有6个样品的均值都几乎在同一条线上,这样的数据可以用于后续的分析。如果不在同一条线上,说明有批次效应,就需要使用limma包内置的normalizeBetweenArrays进行人工校正一下。
library(limma)
exp = normalizeBetweenArrays(exp)
- 管家基因表达量分析
而GAPDH和ACTB的表达量都在14左右,几乎已经是boxplot的上限,所以可以从另一方面说明上面对基因symbol的替换是正确的。
exp['GAPDH',]
exp['ACTB',]

- PCA图-hclust图分析样本分组信息
library(ggfortify)
# 互换行和列
df=as.data.frame(t(exp))
df$group=group_list
autoplot(prcomp( df[,1:(ncol(df)-1)] ), data=df,colour = 'group') # 绘图
在PC1维度,两者可以分的很开,说明两个实验之间确实有差异。符合预期,可以进行后续分析。

# 更改表达矩阵列名
colnames(exp) = paste(group_list,1:6,sep='')
# 定义nodePar
nodePar <- list(lab.cex = 0.6, pch = c(NA, 19), cex = 0.7, col = "blue")
# 聚类
hc=hclust(dist(t(exp)))
par(mar=c(5,5,5,10))
# 绘图
plot(as.dendrogram(hc), nodePar = nodePar, horiz = TRUE)
两者可以分的聚类,说明两个实验之间确实有差异。符合预期,可以进行后续分析。

当把所有的数据都分析好了之后,记得保存表达矩阵的数据:
save(exp,group_list,file = "step2output.Rdata")
差异分析
使用limma包进行差异分析需要三个数据:
- 表达矩阵(已经准备好了,即上面的exp)
- 分组矩阵
- 差异比较矩阵
- 分组矩阵
# rm(list = ls()) ## 一键清空整个环境
options(stringsAsFactors = F)
load(file = "step2output.Rdata")
# 加载limma包
library(limma)
# 做分组矩阵
design <- model.matrix(~0+factor(group_list))
colnames(design)=levels(factor(group_list))
rownames(design)=colnames(exp)
design #分组矩阵 就是这个design变量了
- 差异比较矩阵
contrast.matrix<-makeContrasts(paste0(c("treat","contorl"),collapse = "-"),levels = design)
contrast.matrix # 差异比较矩阵 就是这个了。
使用limma包做差异分析一共就三步,不用问为啥,照着输入就可以了:
- lmFit
- eBayes
- topTable
##step1
fit <- lmFit(exp,design)
##step2
fit2 <- contrasts.fit(fit, contrast.matrix) # 这一步很重要,也就是让软件进行treat/control的操作。
fit2 <- eBayes(fit2)
##step3
tempOutput = topTable(fit2, coef=1, n=Inf)
nrDEG = na.omit(tempOutput)
write.csv(nrDEG,"limma_notrend.results.csv",quote = F) # 把结果写入csv文件
save(nrDEG,file = "DEGoutput.Rdata") # 保存结果
结果可视化
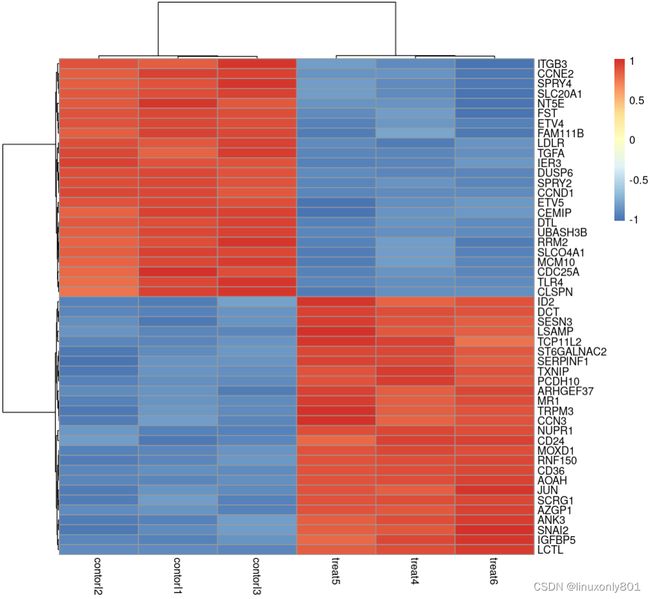
- 热图
library(pheatmap)
choose_gene = head(rownames(nrDEG),50) # 选出最有差异的前50个基因。选择基因的个数看自己心情。
choose_matrix = exp[choose_gene,]
choose_matrix = t(scale(t(choose_matrix))) # 缩放数值到[-1, 1]之间。如果不缩放,就真心没法看了。
pheatmap(choose_matrix)
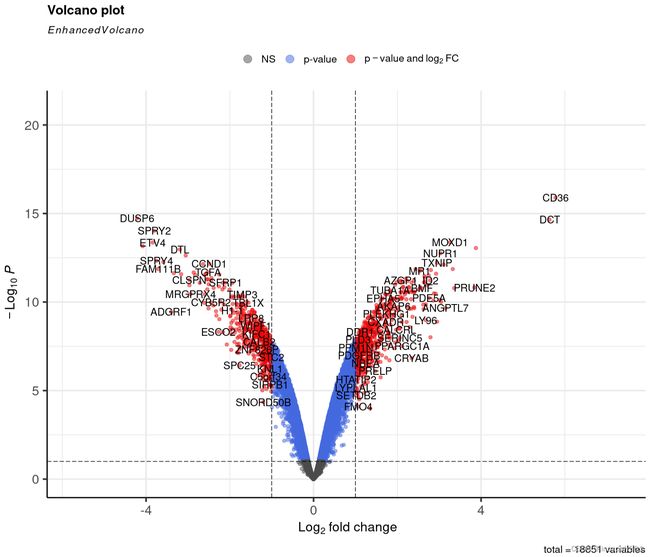
- 火山图
library(EnhancedVolcano)
EnhancedVolcano(nrDEG, lab = rownames(nrDEG), x = 'logFC', y = 'P.Value', pCutoff = 10e-2)
富集分析
KEGG支持的物种列表:https://www.genome.jp/kegg/catalog/org_list.html
GO支持的物种列表:http://bioconductor.org/packages/release/BiocViews.html#___OrgDb
参考:https://zhuanlan.zhihu.com/p/377356510
library(clusterProfiler)
info <- read.csv('limma_notrend.results.csv', header = T)
gene <- bitr(info$X, fromType = 'SYMBOL', toType = 'ENTREZID', OrgDb = 'org.Hs.eg.db')
# info$X中的X是read.csv时没有指定row.names = 1,系统自动分配的列名。
# OrgDb的可选参数在:GO支持的物种列表中可以查看
# GO分析
GO_database <- 'org.Hs.eg.db'
GO<-enrichGO(gene$ENTREZID,
OrgDb = GO_database,
keyType = 'ENTREZID',
ont='ALL',
pvalueCutoff = 0.05,
qvalueCutoff = 0.05,
readable = T)
# KEGG分析
KEGG_database <- 'hsa'
KEGG<-enrichKEGG(gene$ENTREZID,
organism = KEGG_database,
pvalueCutoff = 0.05,
qvalueCutoff = 0.05)
# 可视化作图
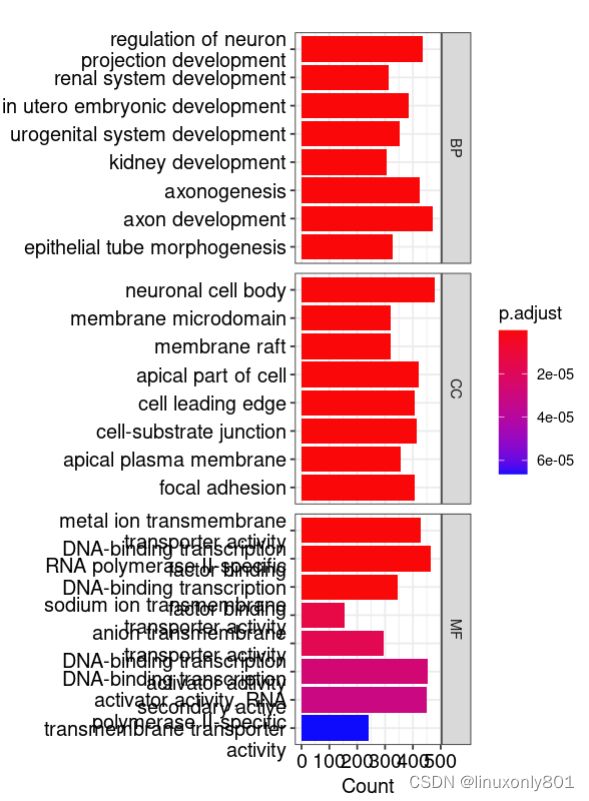
barplot(GO, split='ONTOLOGY') + facet_grid(ONTOLOGY~., scales = 'free') # 柱状图
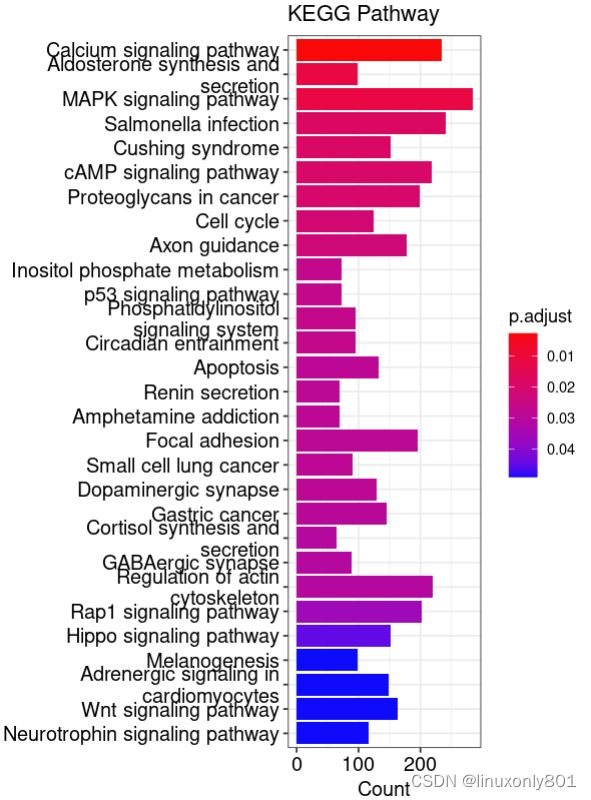
barplot(KEGG,showCategory = 40,title = 'KEGG Pathway')
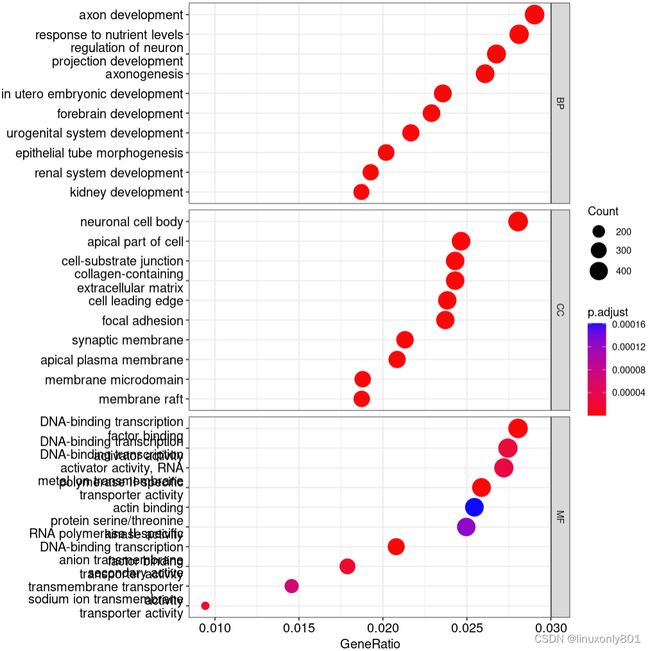
dotplot(GO, split="ONTOLOGY") + facet_grid(ONTOLOGY~., scale='free') # 气泡图
dotplot(KEGG)