参考文献:Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown
流程示意图
安装软件
HISAT2:将reads比对到基因组上
wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/downloads/hisat2-2.1.0-Linux_x86_64.zip
unzip hisat2-2.1.0-Linux_x86_64.zip
echo 'export PATH=/home/yt/biotools/hisat2-2.1.0/bin:$PATH' >> ~/.bashrc
StringTie:将比对好的reads进行拼装并预计表达水平
wget http://ccb.jhu.edu/software/stringtie/dl/stringtie-2.0.4.Linux_x86_64.tar.gz
tar -zvxf stringtie-2.0.4.Linux_x86_64.tar.gz
echo 'export PATH=/home/yt/biotools/stringtie-2.0.4.Linux_x86_64:$PATH' >> ~/.bashrc
SAMtools
sudo apt install samtools
gffcompare:
将基因和转录本与注释进行比较,并报告有关此比较的统计数据,确定组装的转录本是否完全或部分地匹配注释的基因,并计算出多少完全是新的
wget http://ccb.jhu.edu/software/stringtie/dl/gffcompare-0.11.5.Linux_x86_64.tar.gz
tar -zxvf gffcompare-0.11.5.tar.gz
echo 'export PATH=/home/yt/biotools/gffcompare-0.11.5.Linux_x86_64:$PATH' >> ~/.bashrc
ballgown和clusterProfiler(R语言)
ballgown:基因差异表达分析的工具,能利用RNA-Seq实验的数据,预测基因、转录本的差异表达。
clusterProfiler:富集分析工具
source("https://bioconductor.org/biocLite.R") #Bioconductor软件库
biocLite(pkgs = c("ballgown"))
biocLite(pkgs = c("genefilter"))
biocLite(pkgs = c("RSkittleBrewer"))
biocLite(pkgs = c("ggplot2"))
biocLite(pkgs = c("devtools"))
biocLite(pkgs = c("dplyr"))
在R里使用ballgown处理需要得到:
1. 表型数据 关于样本的信息
2. 表达数据 标准化和未标准化的关于外显子,junction,转录本,基因的表达数量
3. 基因信息 有关外显子,junction,转录本,基因的坐标以及注释信息
大多数差异表达分析都会包括一下几个步骤: #需要着重理解
1. 数据可视化和检查
2. 差异表达的统计分析
3. 多重检验校正
4. 下游检查和数据summary
数据 :https://pan.baidu.com/s/1aX93Q65Dit3iqslRWkQcsw
genes 针对基因组的注释文件.gtf
genome 染色体X的序列文件 chrX.fa
geuvadis_phenodata.csv
mergelist.txt 以上两个都是之前博主创建表明数据关系的文件
indexes hisat2对于染色体X的indexes文件,1~8.ht2 索引文件
samples 数据 fastq.gz
分析(提前把文件解压好,数据解压出来&提前装好fastQC,hisat2,stringtie,samtools,R, ballgown)
质控,运行fastQC
nohup fastqc filename.fq & for i in *fastq;
do
i=${i%fastq*};
fastqc ${i}fastq
done
输出文件为filename.fastqc.zip,可在网页上看结果.
HISAT2
建立索引
./extract_splice_sites.py /home/yt/Documents/数据/chrX_data/genes/chrX.gtf>/home/yt/Documents/数据/chrX_data/genes/chrX.ss
./extract_exons.py /home/yt/Documents/数据/chrX_data/genes/chrX.gtf>/home/yt/Documents/数据/chrX_data/genes/chrX.exon
hisat2-build --ss chrX.ss --exon chrX.exon chrX_data / genome / chrX.fa chrX_tran
进行比对
for i in {188044,188104,188234,188245,188257,188273,188337,188383,188401,188428,188454,204916};do hisat2 -p 4 -x chrX_data/indexes/chrX_tran -1 chrX_data/samples/ERR${i}_chrX_1.fastq.gz -2 chrX_data/samples/ERR${i}_chrX_2.fastq.gz -S ERR${i}_chrX.sam done
SAMtools
sam文件转化成bam文件:
samtools view -bS /home/yt/Documents/数据/chrX_data/results/hisat2/ERR188044.sam > /home/yt/Documents/数据/chrX_data/results/stringtie/ERR188044.bam
排序:
samtools sort /home/yt/Documents/数据/chrX_data/results/stringtie/bam/ERR188044.bam /home/yt/Documents/数据/chrX_data/results/stringtie/bam/ERR188044.sort.bam
stringtie
进行拼接:会对每个bam文件生产一个gtf文件,主要记录i转录本的组装信息。
for i in {188044,188104,188234,188245,188257,188273,188337,188383,188401,188428,188454,204916};do stringtie -p 8 -G ./genes/chrX.gtf -o ERR${i}_chrX.gtf -l ERR${i} ERR${i}_chrX.bam done
./stringtie --merge -G /home/yt/Documents/数据/chrX_data/genes/chrX.gtf -o /home/yt/Documents/数据/chrX_data/results/stringtie/stringtie_merged.gtf /home/yt/Documents/数据/chrX_data/results/stringtie/mergelist.txt
再次运行stringtie,组装转录本并估算基因表达丰度:
./stringtie -e -B -G /home/yt/Documents/数据/chrX_data/results/stringtie/stringtie_merged.gtf -o /home/yt/Documents/数据/chrX_data/results/ballgown/ballgown.ERR188383 /home/yt/Documents/数据/chrX_data/results/stringtie/bam/ERR188383.sort.bam
StringTie使用-B参数直接生成Ballgown的输入文件,Cufflinks的输出结果需要使用Tablemaker生成Ballgown的输入文件。
一个有5个输入文件,分别是:
e_data.ctab,外显子水平表达量;
i_data.ctab,内含子水平表达量;
t_data.ctab,转录本水平表达量;
e2t.ctab,外显子与转录本的对应关系;
i2t.ctab,内含子与转录本的对应关系。
进行差异表达分析: ballgown
setwd('/home/yt/Documents/数据/chrX_data')
pheno_data = read.csv("geuvadis_phenodata.csv")
library(ballgown)
library(RSkittleBrewer)
library(genefilter)
library(dplyr)
library(devtools)
bg_chrX = ballgown(dataDir = "/home/yt/Documents/数据/chrX_data/results/ballgown", samplePattern = "ERR", pData=pheno_data)
bg_chrX_filt = subset(bg_chrX,"rowVars(texpr(bg_chrX)) >1",genomesubset=TRUE)
results_transcripts = stattest(bg_chrX_filt,feature="transcript",covariate="sex",adjustvars = c("population"), getFC=TRUE, meas="FPKM")
results_genes = stattest(bg_chrX_filt, feature="gene", covariate="sex", adjustvars = c("population"), getFC=TRUE,meas="FPKM")
results_transcripts = data.frame(geneNames=ballgown::geneNames(bg_chrX_filt), geneIDs=ballgown::geneIDs(bg_chrX_filt), results_transcripts)
results_transcripts = arrange(results_transcripts,pval)
results_genes = arrange(results_genes,pval)
write.csv(results_transcripts, "chrX_transcript_results.csv", row.names=FALSE)
write.csv(results_genes, "chrX_gene_results.csv",row.names=FALSE)
subset(results_transcripts,results_transcripts$qval<0.05)
subset(results_genes,results_genes$qval<0.05)
数据可视化:
#按照p-value从小到大排序
> result_transcripts=arrange(result_transcripts,pval)
> result_genes=arrange(result_genes,pval)
#把两个结果写入到csv文件中
> write.csv(result_transcripts, "chrX_transcript_results.csv",row.names=FALSE)
> write.csv(result_genes, "chrX_gene_results.csv",row.names=FALSE)
#从以上的输出中筛选出q值小于0.05的transcripts和genes,即性别间表达有差异的基因
> subset(result_transcripts,result_transcripts$qval<0.05)
> subset(result_genes,result_genes$qval<0.05)
#赋予调色板五个指定颜色
> tropical= c('darkorange', 'dodgerblue','hotpink', 'limegreen', 'yellow')
> palette(tropical)
#以FPKM为参考值作图,以性别作为区分条件
> fpkm = texpr(bg_chrX,meas="FPKM")
#方便作图将其log转换,+1是为了避免出现log2(0)的情况
> fpkm = log2(fpkm+1)
>boxplot(fpkm,col=as.numeric(pheno_data$sex),las=2,ylab='log2(FPKM+1)')
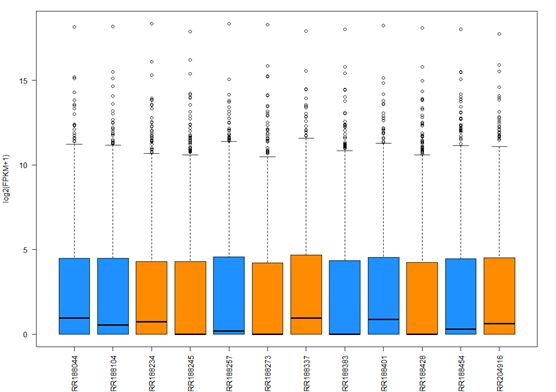
#查看单个转录本在样品中的分布,如图,并绘制箱线图
> ballgown::transcriptNames(bg_chrX)[12]
> ballgown::geneNames(bg_chrX)[12]
>plot(fpkm[12,] ~ pheno_data$sex, border=c(1,2), main=paste(ballgown::geneNames(bg_chrX)[12],' : ', ballgown::transcriptNames(bg_chrX)[12]),pch=19, xlab="Sex", ylab='log2(FPKM+1)')
>points(fpkm[12,] ~ jitter(as.numeric(pheno_data$sex)), col=as.numeric(pheno_data$sex))
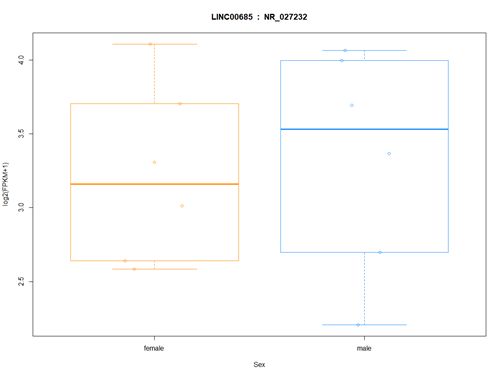
# plotTranscripts函数可以根据指定基因的id画出在特定区段的转录本,可以通过sample函数指定看在某个样本中的表达情况,这里选用id=1750, sample=ERR188234
> plotTranscripts(ballgown::geneIDs(bg_chrX)[1750], bg_chrX, main=c('Gene MSTRG.575 in sample ERR188234'), sample=c('ERR188234'))

# 这里以id=575的基因为例(对应上一步作图)
> plotMeans('MSTRG.575', bg_chrX_filt,groupvar="sex",legend=FALSE)
