最近发现了python版的MCScan,是个大宝藏。由于走了不少弯路,终于画出美图,赶紧记录下来
github地址 https://github.com/tanghaibao/jcvi/wiki/MCscan-(Python-version)
1、软件安装
需要安装LASTAL和jcvi python包
sudo apt install last-align
pip install jcvi
2、输入数据
输入数据只有两类cds和bed文件
可以自动从phytozome,这点十分方便
$ python -m jcvi.apps.fetch phytozome
...
Acoerulea Alyrata Athaliana
Bdistachyon Brapa Cclementina
Cpapaya Creinhardtii Crubella
Csativus Csinensis Csubellipsoidea_C-169
Egrandis Fvesca Gmax
Graimondii Lusitatissimum Mdomestica
Mesculenta Mguttatus Mpusilla_CCMP1545
Mpusilla_RCC299 Mtruncatula Olucimarinus
Osativa Ppatens Ppersica
Ptrichocarpa Pvirgatum Pvulgaris
Rcommunis Sbicolor Sitalica
Slycopersicum Smoellendorffii Stuberosum
Tcacao Thalophila Vcarteri
Vvinifera Zmays early_release
以水稻和拟南芥为例
$ python -m jcvi.apps.fetch phytozome Osativa,Athaliana
$ ls
Athaliana_167_cds.fa.gz Athaliana_167_gene.gff3.gz Osativa_204_cds.fa.gz Osativa_204_gene.gff3.gz
其中gff3文件不需要解压 一键转换成bed格式
python -m jcvi.formats.gff bed --type=mRNA --key=Name Osativa_204_gene.gff3.gz -o osa.bed
cds解压后需要去掉|分隔符 b并要修改id 以基因而不是转录本命名
$ gunzip Athaliana_167_cds.fa.gz
$ mv Athaliana_167_cds.fa ath.cds
$ sed 's/\.*$//g' -i ath.cds #也可以这么做 python -m jcvi.formats.fasta format --sep="|" Athaliana_167_cds.fa.gz ath.cds
$ sed 's/\.//g' -i ath.cds
如果是其他物种或者自己组装的基因组数据,记得基因id需要遵循在染色体上的位置从大到小排序的命名原则,否则软件会在gff3转bed的时候自动命名,务必要和cds里的id对应。
3、Pairwise synteny 分析
$ python -m jcvi.compara.catalog ortholog osa ath
分析过程很快,结果包括.anchors文件,点阵图,如果遇到报错,多半是要安装python包,更新Latex。结果文件的含义“The .last file is raw LAST output, .last.filtered is filtered LAST output, .anchors is the seed synteny blocks (high quality), .lifted.anchors recruits additional anchors to form the final synteny blocks.”
$ ls osa.ath.*
osa.ath.lifted.anchors osa.ath.anchors osa.ath.last.filtered osa.ath.last
4、可视化
重头戏来了
a 共线性图
首先生成.simple文件
python -m jcvi.compara.synteny screen --minspan=30 --simple osa.ath.anchors osa.ath.anchors.new
再编辑两个配置文件seqids和layout
$ vi seqids #设置需要展示等染色体号
Chr1,Chr2,Chr3,Chr4,Chr5,Chr6,Chr7,Chr8,Chr9,Chr10,Chr11,Chr12 #osa
Chr1,Chr2,Chr3,Chr4,Chr5,Chr6,Chr7,Chr8,Chr9,Chr10,Chr11,Chr12 #ath
$ vi layout #设置颜色、长宽等
# y, xstart, xend, rotation, color, label, va, bed
.6, .1, .8, 0, , Osa, top, osa.bed
.4, .1, .8, 0, , Ath, top, ath.bed
# edges
e, 0, 1, osa.ath.anchors.simple
接下来就是见证奇迹的时刻

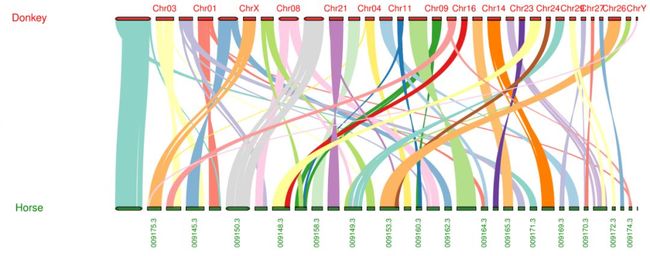
突出显示
$ vi XXX.XXXanchors.simple
g*GSVIVT01012028001 GSVIVT01000604001 ppa011886m ppa008534m 392 +
GSVIVT01010441001 GSVIVT01000970001 ppa022891m ppa001358m 115 -
GSVIVT01000555001 GSVIVT01003228001 ppa002809m ppa010569m 359 +
...
$ python -m jcvi.graphics.karyotype seqids layout

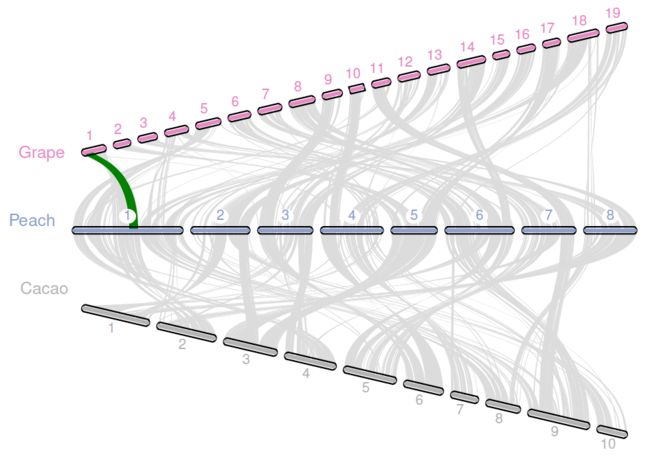
$ vi layout
# y, xstart, xend, rotation, color, label, va, bed
.7, .1, .8, 15, , Grape, top, grape.bed
.5, .1, .8, 0, , Peach, top, peach.bed
.3, .1, .8, -15, , Cacao, bottom, cacao.bed
# edges
e, 0, 1, grape.peach.anchors.simple
e, 1, 2, peach.cacao.anchors.simple
$ vi seqids
chr1,chr2,chr3,chr4,chr5,chr6,chr7,chr8,chr9,chr10,chr11,chr12,chr13,chr14,chr15,chr16,chr17,chr18,chr19
scaffold_1,scaffold_2,scaffold_3,scaffold_4,scaffold_5,scaffold_6,scaffold_7,scaffold_8
scaffold_1,scaffold_2,scaffold_3,scaffold_4,scaffold_5,scaffold_6,scaffold_7,scaffold_8,scaffold_9,scaffold_10r
$ python -m jcvi.graphics.karyotype seqids layout


b dotplot
亲测点阵图是自动出来的,当然也可以用命令行
$ python -m jcvi.graphics.dotplot osa.ath.anchors


查看synteny depth分布
python -m jcvi.compara.synteny depth --histogram osa.ath.anchors

anyway,先介绍到这里啦
更多请参考
基因组共线性工具MCScanX使用说明
基因组间共线性分析想学吗?
无限个!物种共线性分析结果可视化