- 情绪觉察日记第37天
露露_e800
今天是家庭关系规划师的第二阶最后一天,慧萍老师帮我做了个案,帮我处理了埋在心底好多年的一份恐惧,并给了我深深的力量!这几天出来学习,爸妈过来婆家帮我带小孩,妈妈出于爱帮我收拾东西,并跟我先生和婆婆产生矛盾,妈妈觉得他们没有照顾好我…。今晚回家见到妈妈,我很欣赏她并赞扬她,妈妈说今晚要跟我睡我说好,当我们俩躺在床上准备睡觉的时候,我握着妈妈的手对她说:妈妈这几天辛苦你了,你看你多利害把我们的家收拾得
- 机器学习与深度学习间关系与区别
ℒℴѵℯ心·动ꦿ໊ོ꫞
人工智能学习深度学习python
一、机器学习概述定义机器学习(MachineLearning,ML)是一种通过数据驱动的方法,利用统计学和计算算法来训练模型,使计算机能够从数据中学习并自动进行预测或决策。机器学习通过分析大量数据样本,识别其中的模式和规律,从而对新的数据进行判断。其核心在于通过训练过程,让模型不断优化和提升其预测准确性。主要类型1.监督学习(SupervisedLearning)监督学习是指在训练数据集中包含输入
- 铭刻于星(四十二)
随风至
69夜晚,绍敏同学做完功课后,看了眼房外,没听到动静才敢从书包的夹层里拿出那个心形纸团。折痕压得很深,都有些旧了,想来是已经写好很久了。绍敏同学慢慢地、轻轻地捏开折叠处,待到全部拆开后,又反复抚平纸张,然后仔细地一字字默看。只是开头的三个字是第一次看到,让她心漏跳了几拍。“亲爱的绍敏:从四年级的时候,我就喜欢你了,但是我一直不敢说,怕影响你学习。六年级的时候听说有人跟你表白,你接受了,我很难过,但
- UI学习——cell的复用和自定义cell
Magnetic_h
ui学习
目录cell的复用手动(非注册)自动(注册)自定义cellcell的复用在iOS开发中,单元格复用是一种提高表格(UITableView)和集合视图(UICollectionView)滚动性能的技术。当一个UITableViewCell或UICollectionViewCell首次需要显示时,如果没有可复用的单元格,则视图会创建一个新的单元格。一旦这个单元格滚动出屏幕,它就不会被销毁。相反,它被添
- 10月|愿你的青春不负梦想-读书笔记-01
Tracy的小书斋
本书的作者是俞敏洪,大家都很熟悉他了吧。俞敏洪老师是我行业的领头羊吧,也是我事业上的偶像。本日摘录他书中第一章中的金句:『一个人如果什么目标都没有,就会浑浑噩噩,感觉生命中缺少能量。能给我们能量的,是对未来的期待。第一件事,我始终为了进步而努力。与其追寻全世界的骏马,不如种植丰美的草原,到时骏马自然会来。第二件事,我始终有阶段性的目标。什么东西能给我能量?答案是对未来的期待。』读到这里的时候,我便
- 理解Gunicorn:Python WSGI服务器的基石
范范0825
ipythonlinux运维
理解Gunicorn:PythonWSGI服务器的基石介绍Gunicorn,全称GreenUnicorn,是一个为PythonWSGI(WebServerGatewayInterface)应用设计的高效、轻量级HTTP服务器。作为PythonWeb应用部署的常用工具,Gunicorn以其高性能和易用性著称。本文将介绍Gunicorn的基本概念、安装和配置,帮助初学者快速上手。1.什么是Gunico
- 学点心理知识,呵护孩子健康
静候花开_7090
昨天听了华中师范大学教育管理学系副教授张玲老师的《哪里才是学生心理健康的最后庇护所,超越教育与技术的思考》的讲座。今天又重新学习了一遍,收获匪浅。张玲博士也注意到了当今社会上的孩子由于心理问题导致的自残、自杀及伤害他人等恶性事件。她向我们普及了一个重要的命题,她说心理健康的一些基本命题,我们与我们通常的一些教育命题是不同的,她还举了几个例子,让我们明白我们原来以为的健康并非心理学上的健康。比如如果
- 《投行人生》读书笔记
小蘑菇的树洞
《投行人生》----作者詹姆斯-A-朗德摩根斯坦利副主席40年的职业洞见-很短小精悍的篇幅,比较适合初入职场的新人。第一部分成功的职业生涯需要规划1.情商归为适应能力分享与协作同理心适应能力,更多的是自我意识,你有能力识别自己的情并分辨这些情绪如何影响你的思想和行为。2.对于初入职场的人的建议,细节,截止日期和数据很重要截止日期,一种有效的方法是请老板为你所有的任务进行优先级排序。和老板喝咖啡的好
- ArcGIS栅格计算器常见公式(赋值、0和空值的转换、补充栅格空值)
研学随笔
arcgis经验分享
我们在使用ArcGIS时通常经常用到栅格计算器,今天主要给大家介绍我日常中经常用到的几个公式,供大家参考学习。将特定值(-9999)赋值为0,例如-9999.Con("raster"==-9999,0,"raster")2.给空值赋予特定的值(如0)Con(IsNull("raster"),0,"raster")3.将特定的栅格值(如1)赋值为空值,其他保留原值SetNull("raster"==
- 【一起学Rust | 设计模式】习惯语法——使用借用类型作为参数、格式化拼接字符串、构造函数
广龙宇
一起学Rust#Rust设计模式rust设计模式开发语言
提示:文章写完后,目录可以自动生成,如何生成可参考右边的帮助文档文章目录前言一、使用借用类型作为参数二、格式化拼接字符串三、使用构造函数总结前言Rust不是传统的面向对象编程语言,它的所有特性,使其独一无二。因此,学习特定于Rust的设计模式是必要的。本系列文章为作者学习《Rust设计模式》的学习笔记以及自己的见解。因此,本系列文章的结构也与此书的结构相同(后续可能会调成结构),基本上分为三个部分
- 回溯 Leetcode 332 重新安排行程
mmaerd
Leetcode刷题学习记录leetcode算法职场和发展
重新安排行程Leetcode332学习记录自代码随想录给你一份航线列表tickets,其中tickets[i]=[fromi,toi]表示飞机出发和降落的机场地点。请你对该行程进行重新规划排序。所有这些机票都属于一个从JFK(肯尼迪国际机场)出发的先生,所以该行程必须从JFK开始。如果存在多种有效的行程,请你按字典排序返回最小的行程组合。例如,行程[“JFK”,“LGA”]与[“JFK”,“LGB
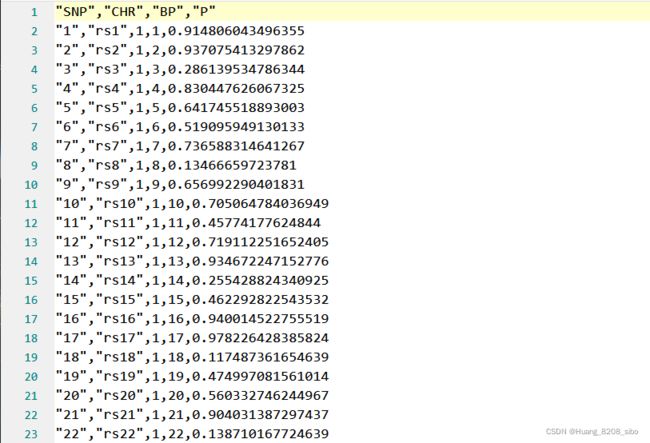
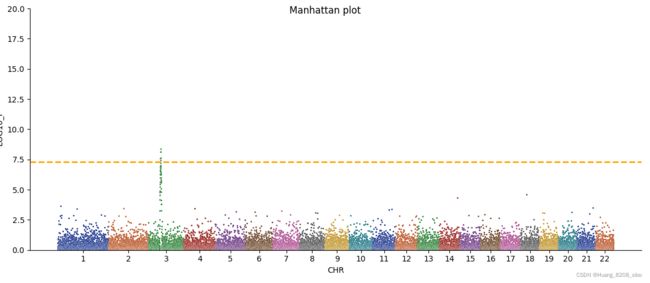
- Python数据分析与可视化实战指南
William数据分析
pythonpython数据
在数据驱动的时代,Python因其简洁的语法、强大的库生态系统以及活跃的社区,成为了数据分析与可视化的首选语言。本文将通过一个详细的案例,带领大家学习如何使用Python进行数据分析,并通过可视化来直观呈现分析结果。一、环境准备1.1安装必要库在开始数据分析和可视化之前,我们需要安装一些常用的库。主要包括pandas、numpy、matplotlib和seaborn等。这些库分别用于数据处理、数学
- git常用命令笔记
咩酱-小羊
git笔记
###用习惯了idea总是不记得git的一些常见命令,需要用到的时候总是担心旁边站了人~~~记个笔记@_@,告诉自己看笔记不丢人初始化初始化一个新的Git仓库gitinit配置配置用户信息gitconfig--globaluser.name"YourName"gitconfig--globaluser.email"
[email protected]"基本操作克隆远程仓库gitclone查看
- python os.environ
江湖偌大
python深度学习
os.environ['TF_CPP_MIN_LOG_LEVEL']='0'#默认值,输出所有信息os.environ['TF_CPP_MIN_LOG_LEVEL']='1'#屏蔽通知信息(INFO)os.environ['TF_CPP_MIN_LOG_LEVEL']='2'#屏蔽通知信息和警告信息(INFO\WARNING)os.environ['TF_CPP_MIN_LOG_LEVEL']='
- Python中os.environ基本介绍及使用方法
鹤冲天Pro
#Pythonpython服务器开发语言
文章目录python中os.environos.environ简介os.environ进行环境变量的增删改查python中os.environ的使用详解1.简介2.key字段详解2.1常见key字段3.os.environ.get()用法4.环境变量的增删改查和判断是否存在4.1新增环境变量4.2更新环境变量4.3获取环境变量4.4删除环境变量4.5判断环境变量是否存在python中os.envi
- Pyecharts数据可视化大屏:打造沉浸式数据分析体验
我的运维人生
信息可视化数据分析数据挖掘运维开发技术共享
Pyecharts数据可视化大屏:打造沉浸式数据分析体验在当今这个数据驱动的时代,如何将海量数据以直观、生动的方式展现出来,成为了数据分析师和企业决策者关注的焦点。Pyecharts,作为一款基于Python的开源数据可视化库,凭借其丰富的图表类型、灵活的配置选项以及高度的定制化能力,成为了构建数据可视化大屏的理想选择。本文将深入探讨如何利用Pyecharts打造数据可视化大屏,并通过实际代码案例
- 2019-12-22-22:30
涓涓1016
今天是冬至,写下我的日更,是因为这两天的学习真的是能量的满满,让我看到了自己,未来另外一种可能性,也让我看到了这两年这几年的过程中我所接受那些痛苦的来源。一切的根源和痛苦都来自于人生,家庭,而你的原生家庭,你的爸爸和妈妈,是因为你这个灵魂在那一刻选择他们作为你的爸爸和妈妈来的,所以你得接受他,你得接纳他,他就是因为他的存在而给你的学习和成长带来这些痛苦,那其实是你必然要经历的这个过程,当你去接纳的
- Python教程:一文了解使用Python处理XPath
旦莫
Python进阶python开发语言
目录1.环境准备1.1安装lxml1.2验证安装2.XPath基础2.1什么是XPath?2.2XPath语法2.3示例XML文档3.使用lxml解析XML3.1解析XML文档3.2查看解析结果4.XPath查询4.1基本路径查询4.2使用属性查询4.3查询多个节点5.XPath的高级用法5.1使用逻辑运算符5.2使用函数6.实战案例6.1从网页抓取数据6.1.1安装Requests库6.1.2代
- python os.environ_python os.environ 读取和设置环境变量
weixin_39605414
pythonos.environ
>>>importos>>>os.environ.keys()['LC_NUMERIC','GOPATH','GOROOT','GOBIN','LESSOPEN','SSH_CLIENT','LOGNAME','USER','HOME','LC_PAPER','PATH','DISPLAY','LANG','TERM','SHELL','J2REDIR','LC_MONETARY','QT_QPA
- 将cmd中命令输出保存为txt文本文件
落难Coder
Windowscmdwindow
最近深度学习本地的训练中我们常常要在命令行中运行自己的代码,无可厚非,我们有必要保存我们的炼丹结果,但是复制命令行输出到txt是非常麻烦的,其实Windows下的命令行为我们提供了相应的操作。其基本的调用格式就是:运行指令>输出到的文件名称或者具体保存路径测试下,我打开cmd并且ping一下百度:pingwww.baidu.com>./data.txt看下相同目录下data.txt的输出:如果你再
- 509. 斐波那契数(每日一题)
lzyprime
lzyprime博客(github)创建时间:2021.01.04qq及邮箱:2383518170leetcode笔记题目描述斐波那契数,通常用F(n)表示,形成的序列称为斐波那契数列。该数列由0和1开始,后面的每一项数字都是前面两项数字的和。也就是:F(0)=0,F(1)=1F(n)=F(n-1)+F(n-2),其中n>1给你n,请计算F(n)。示例1:输入:2输出:1解释:F(2)=F(1)+
- 拥有断舍离的心态,过精简生活--《断舍离》读书笔记
爱吃丸子的小樱桃
不知不觉间房间里的东西越来越多,虽然摆放整齐,但也时常会觉得空间逼仄,令人心生烦闷。抱着断舍离的态度,我开始阅读《断舍离》这本书,希望从书中能找到一些有效的方法,帮助我实现空间、物品上的断舍离。《断舍离》是日本作家山下英子通过自己的经历、思考和实践总结而成的,整体内涵也从刚开始的私人生活哲学的“断舍离”升华成了“人生实践哲学”,接着又成为每个人都能实行的“改变人生的断舍离”,从“哲学”逐渐升华成“
- 四章-32-点要素的聚合
彩云飘过
本文基于腾讯课堂老胡的课《跟我学Openlayers--基础实例详解》做的学习笔记,使用的openlayers5.3.xapi。源码见1032.html,对应的官网示例https://openlayers.org/en/latest/examples/cluster.htmlhttps://openlayers.org/en/latest/examples/earthquake-clusters.
- 高端密码学院笔记285
柚子_b4b4
高端幸福密码学院(高级班)幸福使者:李华第(598)期《幸福》之回归内在深层生命原动力基础篇——揭秘“激励”成长的喜悦心理案例分析主讲:刘莉一,知识扩充:成功=艰苦劳动+正确方法+少说空话。贪图省力的船夫,目标永远下游。智者的梦再美,也不如愚人实干的脚印。幸福早课堂2020.10.16星期五一笔记:1,重视和珍惜的前提是知道它的价值非常重要,当你珍惜了,你就真正定下来,真正的学到身上。2,大家需要
- 使用Faiss进行高效相似度搜索
llzwxh888
faisspython
在现代AI应用中,快速和高效的相似度搜索是至关重要的。Faiss(FacebookAISimilaritySearch)是一个专门用于快速相似度搜索和聚类的库,特别适用于高维向量。本文将介绍如何使用Faiss来进行相似度搜索,并结合Python代码演示其基本用法。什么是Faiss?Faiss是一个由FacebookAIResearch团队开发的开源库,主要用于高维向量的相似性搜索和聚类。Faiss
- python是什么意思中文-在python中%是什么意思
编程大乐趣
Python中%有两种:1、数值运算:%代表取模,返回除法的余数。如:>>>7%212、%操作符(字符串格式化,stringformatting),说明如下:%[(name)][flags][width].[precision]typecode(name)为命名flags可以有+,-,''或0。+表示右对齐。-表示左对齐。''为一个空格,表示在正数的左侧填充一个空格,从而与负数对齐。0表示使用0填
- GitHub上克隆项目
bigbig猩猩
github
从GitHub上克隆项目是一个简单且直接的过程,它允许你将远程仓库中的项目复制到你的本地计算机上,以便进行进一步的开发、测试或学习。以下是一个详细的步骤指南,帮助你从GitHub上克隆项目。一、准备工作1.安装Git在克隆GitHub项目之前,你需要在你的计算机上安装Git工具。Git是一个开源的分布式版本控制系统,用于跟踪和管理代码变更。你可以从Git的官方网站(https://git-scm.
- HTML网页设计制作大作业(div+css) 云南我的家乡旅游景点 带文字滚动
二挡起步
web前端期末大作业web设计网页规划与设计htmlcssjavascriptdreamweaver前端
Web前端开发技术描述网页设计题材,DIV+CSS布局制作,HTML+CSS网页设计期末课程大作业游景点介绍|旅游风景区|家乡介绍|等网站的设计与制作HTML期末大学生网页设计作业HTML:结构CSS:样式在操作方面上运用了html5和css3,采用了div+css结构、表单、超链接、浮动、绝对定位、相对定位、字体样式、引用视频等基础知识JavaScript:做与用户的交互行为文章目录前端学习路线
- Day17笔记-高阶函数
~在杰难逃~
Python笔记python开发语言pycharm数据分析
高阶函数【重点掌握】函数的本质:函数是一个变量,函数名是一个变量名,一个函数可以作为另一个函数的参数或返回值使用如果A函数作为B函数的参数,B函数调用完成之后,会得到一个结果,则B函数被称为高阶函数常用的高阶函数:map(),reduce(),filter(),sorted()1.map()map(func,iterable),返回值是一个iterator【容器,迭代器】func:函数iterab
- Day1笔记-Python简介&标识符和关键字&输入输出
~在杰难逃~
Pythonpython开发语言大数据数据分析数据挖掘
大家好,从今天开始呢,杰哥开展一个新的专栏,当然,数据分析部分也会不定时更新的,这个新的专栏主要是讲解一些Python的基础语法和知识,帮助0基础的小伙伴入门和学习Python,感兴趣的小伙伴可以开始认真学习啦!一、Python简介【了解】1.计算机工作原理编程语言就是用来定义计算机程序的形式语言。我们通过编程语言来编写程序代码,再通过语言处理程序执行向计算机发送指令,让计算机完成对应的工作,编程
- 项目中 枚举与注解的结合使用
飞翔的马甲
javaenumannotation
前言:版本兼容,一直是迭代开发头疼的事,最近新版本加上了支持新题型,如果新创建一份问卷包含了新题型,那旧版本客户端就不支持,如果新创建的问卷不包含新题型,那么新旧客户端都支持。这里面我们通过给问卷类型枚举增加自定义注解的方式完成。顺便巩固下枚举与注解。
一、枚举
1.在创建枚举类的时候,该类已继承java.lang.Enum类,所以自定义枚举类无法继承别的类,但可以实现接口。
- 【Scala十七】Scala核心十一:下划线_的用法
bit1129
scala
下划线_在Scala中广泛应用,_的基本含义是作为占位符使用。_在使用时是出问题非常多的地方,本文将不断完善_的使用场景以及所表达的含义
1. 在高阶函数中使用
scala> val list = List(-3,8,7,9)
list: List[Int] = List(-3, 8, 7, 9)
scala> list.filter(_ > 7)
r
- web缓存基础:术语、http报头和缓存策略
dalan_123
Web
对于很多人来说,去访问某一个站点,若是该站点能够提供智能化的内容缓存来提高用户体验,那么最终该站点的访问者将络绎不绝。缓存或者对之前的请求临时存储,是http协议实现中最核心的内容分发策略之一。分发路径中的组件均可以缓存内容来加速后续的请求,这是受控于对该内容所声明的缓存策略。接下来将讨web内容缓存策略的基本概念,具体包括如如何选择缓存策略以保证互联网范围内的缓存能够正确处理的您的内容,并谈论下
- crontab 问题
周凡杨
linuxcrontabunix
一: 0481-079 Reached a symbol that is not expected.
背景:
*/5 * * * * /usr/IBMIHS/rsync.sh
- 让tomcat支持2级域名共享session
g21121
session
tomcat默认情况下是不支持2级域名共享session的,所有有些情况下登陆后从主域名跳转到子域名会发生链接session不相同的情况,但是只需修改几处配置就可以了。
打开tomcat下conf下context.xml文件
找到Context标签,修改为如下内容
如果你的域名是www.test.com
<Context sessionCookiePath="/path&q
- web报表工具FineReport常用函数的用法总结(数学和三角函数)
老A不折腾
Webfinereport总结
ABS
ABS(number):返回指定数字的绝对值。绝对值是指没有正负符号的数值。
Number:需要求出绝对值的任意实数。
示例:
ABS(-1.5)等于1.5。
ABS(0)等于0。
ABS(2.5)等于2.5。
ACOS
ACOS(number):返回指定数值的反余弦值。反余弦值为一个角度,返回角度以弧度形式表示。
Number:需要返回角
- linux 启动java进程 sh文件
墙头上一根草
linuxshelljar
#!/bin/bash
#初始化服务器的进程PId变量
user_pid=0;
robot_pid=0;
loadlort_pid=0;
gateway_pid=0;
#########
#检查相关服务器是否启动成功
#说明:
#使用JDK自带的JPS命令及grep命令组合,准确查找pid
#jps 加 l 参数,表示显示java的完整包路径
#使用awk,分割出pid
- 我的spring学习笔记5-如何使用ApplicationContext替换BeanFactory
aijuans
Spring 3 系列
如何使用ApplicationContext替换BeanFactory?
package onlyfun.caterpillar.device;
import org.springframework.beans.factory.BeanFactory;
import org.springframework.beans.factory.xml.XmlBeanFactory;
import
- Linux 内存使用方法详细解析
annan211
linux内存Linux内存解析
来源 http://blog.jobbole.com/45748/
我是一名程序员,那么我在这里以一个程序员的角度来讲解Linux内存的使用。
一提到内存管理,我们头脑中闪出的两个概念,就是虚拟内存,与物理内存。这两个概念主要来自于linux内核的支持。
Linux在内存管理上份为两级,一级是线性区,类似于00c73000-00c88000,对应于虚拟内存,它实际上不占用
- 数据库的单表查询常用命令及使用方法(-)
百合不是茶
oracle函数单表查询
创建数据库;
--建表
create table bloguser(username varchar2(20),userage number(10),usersex char(2));
创建bloguser表,里面有三个字段
&nbs
- 多线程基础知识
bijian1013
java多线程threadjava多线程
一.进程和线程
进程就是一个在内存中独立运行的程序,有自己的地址空间。如正在运行的写字板程序就是一个进程。
“多任务”:指操作系统能同时运行多个进程(程序)。如WINDOWS系统可以同时运行写字板程序、画图程序、WORD、Eclipse等。
线程:是进程内部单一的一个顺序控制流。
线程和进程
a. 每个进程都有独立的
- fastjson简单使用实例
bijian1013
fastjson
一.简介
阿里巴巴fastjson是一个Java语言编写的高性能功能完善的JSON库。它采用一种“假定有序快速匹配”的算法,把JSON Parse的性能提升到极致,是目前Java语言中最快的JSON库;包括“序列化”和“反序列化”两部分,它具备如下特征:
- 【RPC框架Burlap】Spring集成Burlap
bit1129
spring
Burlap和Hessian同属于codehaus的RPC调用框架,但是Burlap已经几年不更新,所以Spring在4.0里已经将Burlap的支持置为Deprecated,所以在选择RPC框架时,不应该考虑Burlap了。
这篇文章还是记录下Burlap的用法吧,主要是复制粘贴了Hessian与Spring集成一文,【RPC框架Hessian四】Hessian与Spring集成
- 【Mahout一】基于Mahout 命令参数含义
bit1129
Mahout
1. mahout seqdirectory
$ mahout seqdirectory
--input (-i) input Path to job input directory(原始文本文件).
--output (-o) output The directory pathna
- linux使用flock文件锁解决脚本重复执行问题
ronin47
linux lock 重复执行
linux的crontab命令,可以定时执行操作,最小周期是每分钟执行一次。关于crontab实现每秒执行可参考我之前的文章《linux crontab 实现每秒执行》现在有个问题,如果设定了任务每分钟执行一次,但有可能一分钟内任务并没有执行完成,这时系统会再执行任务。导致两个相同的任务在执行。
例如:
<?
//
test
.php
- java-74-数组中有一个数字出现的次数超过了数组长度的一半,找出这个数字
bylijinnan
java
public class OcuppyMoreThanHalf {
/**
* Q74 数组中有一个数字出现的次数超过了数组长度的一半,找出这个数字
* two solutions:
* 1.O(n)
* see <beauty of coding>--每次删除两个不同的数字,不改变数组的特性
* 2.O(nlogn)
* 排序。中间
- linux 系统相关命令
candiio
linux
系统参数
cat /proc/cpuinfo cpu相关参数
cat /proc/meminfo 内存相关参数
cat /proc/loadavg 负载情况
性能参数
1)top
M:按内存使用排序
P:按CPU占用排序
1:显示各CPU的使用情况
k:kill进程
o:更多排序规则
回车:刷新数据
2)ulimit
ulimit -a:显示本用户的系统限制参
- [经营与资产]保持独立性和稳定性对于软件开发的重要意义
comsci
软件开发
一个软件的架构从诞生到成熟,中间要经过很多次的修正和改造
如果在这个过程中,外界的其它行业的资本不断的介入这种软件架构的升级过程中
那么软件开发者原有的设计思想和开发路线
- 在CentOS5.5上编译OpenJDK6
Cwind
linuxOpenJDK
几番周折终于在自己的CentOS5.5上编译成功了OpenJDK6,将编译过程和遇到的问题作一简要记录,备查。
0. OpenJDK介绍
OpenJDK是Sun(现Oracle)公司发布的基于GPL许可的Java平台的实现。其优点:
1、它的核心代码与同时期Sun(-> Oracle)的产品版基本上是一样的,血统纯正,不用担心性能问题,也基本上没什么兼容性问题;(代码上最主要的差异是
- java乱码问题
dashuaifu
java乱码问题js中文乱码
swfupload上传文件参数值为中文传递到后台接收中文乱码 在js中用setPostParams({"tag" : encodeURI( document.getElementByIdx_x("filetag").value,"utf-8")});
然后在servlet中String t
- cygwin很多命令显示command not found的解决办法
dcj3sjt126com
cygwin
cygwin很多命令显示command not found的解决办法
修改cygwin.BAT文件如下
@echo off
D:
set CYGWIN=tty notitle glob
set PATH=%PATH%;d:\cygwin\bin;d:\cygwin\sbin;d:\cygwin\usr\bin;d:\cygwin\usr\sbin;d:\cygwin\us
- [介绍]从 Yii 1.1 升级
dcj3sjt126com
PHPyii2
2.0 版框架是完全重写的,在 1.1 和 2.0 两个版本之间存在相当多差异。因此从 1.1 版升级并不像小版本间的跨越那么简单,通过本指南你将会了解两个版本间主要的不同之处。
如果你之前没有用过 Yii 1.1,可以跳过本章,直接从"入门篇"开始读起。
请注意,Yii 2.0 引入了很多本章并没有涉及到的新功能。强烈建议你通读整部权威指南来了解所有新特性。这样有可能会发
- Linux SSH免登录配置总结
eksliang
ssh-keygenLinux SSH免登录认证Linux SSH互信
转载请出自出处:http://eksliang.iteye.com/blog/2187265 一、原理
我们使用ssh-keygen在ServerA上生成私钥跟公钥,将生成的公钥拷贝到远程机器ServerB上后,就可以使用ssh命令无需密码登录到另外一台机器ServerB上。
生成公钥与私钥有两种加密方式,第一种是
- 手势滑动销毁Activity
gundumw100
android
老是效仿ios,做android的真悲催!
有需求:需要手势滑动销毁一个Activity
怎么办尼?自己写?
不用~,网上先问一下百度。
结果:
http://blog.csdn.net/xiaanming/article/details/20934541
首先将你需要的Activity继承SwipeBackActivity,它会在你的布局根目录新增一层SwipeBackLay
- JavaScript变换表格边框颜色
ini
JavaScripthtmlWebhtml5css
效果查看:http://hovertree.com/texiao/js/2.htm代码如下,保存到HTML文件也可以查看效果:
<html>
<head>
<meta charset="utf-8">
<title>表格边框变换颜色代码-何问起</title>
</head>
<body&
- Kafka Rest : Confluent
kane_xie
kafkaRESTconfluent
最近拿到一个kafka rest的需求,但kafka暂时还没有提供rest api(应该是有在开发中,毕竟rest这么火),上网搜了一下,找到一个Confluent Platform,本文简单介绍一下安装。
这里插一句,给大家推荐一个九尾搜索,原名叫谷粉SOSO,不想fanqiang谷歌的可以用这个。以前在外企用谷歌用习惯了,出来之后用度娘搜技术问题,那匹配度简直感人。
环境声明:Ubu
- Calender不是单例
men4661273
单例Calender
在我们使用Calender的时候,使用过Calendar.getInstance()来获取一个日期类的对象,这种方式跟单例的获取方式一样,那么它到底是不是单例呢,如果是单例的话,一个对象修改内容之后,另外一个线程中的数据不久乱套了吗?从试验以及源码中可以得出,Calendar不是单例。
测试:
Calendar c1 =
- 线程内存和主内存之间联系
qifeifei
java thread
1, java多线程共享主内存中变量的时候,一共会经过几个阶段,
lock:将主内存中的变量锁定,为一个线程所独占。
unclock:将lock加的锁定解除,此时其它的线程可以有机会访问此变量。
read:将主内存中的变量值读到工作内存当中。
load:将read读取的值保存到工作内存中的变量副本中。
- schedule和scheduleAtFixedRate
tangqi609567707
javatimerschedule
原文地址:http://blog.csdn.net/weidan1121/article/details/527307
import java.util.Timer;import java.util.TimerTask;import java.util.Date;
/** * @author vincent */public class TimerTest {
- erlang 部署
wudixiaotie
erlang
1.如果在启动节点的时候报这个错 :
{"init terminating in do_boot",{'cannot load',elf_format,get_files}}
则需要在reltool.config中加入
{app, hipe, [{incl_cond, exclude}]},
2.当generate时,遇到:
ERROR