2021SC@SDUSC基于人工智能的多肽药物分析问题(十三)
基于人工智能的多肽药物分析问题(十三)
2021SC@SDUSC
1. 前言
代码分析已临近尾声了,目前还剩下e2e模式的预测代码,由于两种模式的代码存在部分重叠,所以接下来的代码可能会略过一些重复代码,特此声明
2. 代码分析
if __name__ == "__main__":
args = get_args()
FFDB=args.db
FFindexDB = namedtuple("FFindexDB", "index, data")
ffdb = FFindexDB(read_index(FFDB+'_pdb.ffindex'),
read_data(FFDB+'_pdb.ffdata'))
if not os.path.exists("%s.npz"%args.out_prefix):
pred = Predictor(model_dir=args.model_dir, use_cpu=args.use_cpu)
pred.predict(args.a3m_fn, args.out_prefix, args.hhr, args.atab)
这部分内容与pyrosetta模式的一致,包括了get_args参数准备过程,namedtuple生成具名元组过程,再之后调用了Predictor类的predict方法来进行e2e模式的预测
class Predictor():
def __init__(self, model_dir=None, use_cpu=False):
if model_dir == None:
self.model_dir = "%s/models"%(os.path.dirname(os.path.realpath(__file__)))
else:
self.model_dir = model_dir
#
# define model name
self.model_name = "RoseTTAFold"
if torch.cuda.is_available() and (not use_cpu):
self.device = torch.device("cuda")
else:
self.device = torch.device("cpu")
self.active_fn = nn.Softmax(dim=1)
# define model & load model
self.model = RoseTTAFoldModule_e2e(**MODEL_PARAM).to(self.device)
在Predictor类的构造函数中,同样也是对硬件进行了一系列的判断配置,
- 自动检测电脑的cuda是否能使用,若能够使用并且命令行未指定使用cpu跑模型,那么则利用cuda跑模型。
- 设置了模型的激活函数active function 为Softmax,维度为1
- 加载自定义的RoseTTAFoldModule模型
唯一与之前不同之处在于该模式下,Predictor实例化的模型为RoseTTAFoldModule_e2e,即e2e专用的一个模型。之后调用load_model方法检测相关文件路径能否正确加载数据,是则继续运行,否则结束运行
之后我们重点观察一下e2e的RoseTTAFoldModule的不一样的地方
2.1 RoseTTAFoldModule_e2e模型
REF_param = {
"num_layers" : 3,
"num_channels" : 32,
"num_degrees" : 3,
"l0_in_features": 32,
"l0_out_features": 8,
"l1_in_features": 3,
"l1_out_features": 3,
"num_edge_features": 32,
"div": 4,
"n_heads": 4
}
MODEL_PARAM['SE3_param'] = SE3_param
MODEL_PARAM['REF_param'] = REF_param
在传入该模型的参数方面,e2e相比于pyrosetta的参数中添加了REF_param,下面是RoseTTAFoldModule_e2e模型的构造方法:
class RoseTTAFoldModule_e2e(nn.Module):
def __init__(self, n_module=4, n_module_str=4, n_module_ref=4, n_layer=4,\
d_msa=64, d_pair=128, d_templ=64,\
n_head_msa=4, n_head_pair=8, n_head_templ=4,
d_hidden=64, r_ff=4, n_resblock=1, p_drop=0.0,
performer_L_opts=None, performer_N_opts=None,
SE3_param={'l0_in_features':32, 'l0_out_features':16, 'num_edge_features':32},
REF_param={'l0_in_features':32, 'l0_out_features':16, 'num_edge_features':32},
use_templ=False):
super(RoseTTAFoldModule_e2e, self).__init__()
self.use_templ = use_templ
#
self.msa_emb = MSA_emb(d_model=d_msa, p_drop=p_drop, max_len=5000)
if use_templ:
self.templ_emb = Templ_emb(d_templ=d_templ, n_att_head=n_head_templ, r_ff=r_ff,
performer_opts=performer_L_opts, p_drop=0.0)
self.pair_emb = Pair_emb_w_templ(d_model=d_pair, d_templ=d_templ, p_drop=p_drop)
else:
self.pair_emb = Pair_emb_wo_templ(d_model=d_pair, p_drop=p_drop)
#
self.feat_extractor = IterativeFeatureExtractor(n_module=n_module,\
n_module_str=n_module_str,\
n_layer=n_layer,\
d_msa=d_msa, d_pair=d_pair, d_hidden=d_hidden,\
n_head_msa=n_head_msa, \
n_head_pair=n_head_pair,\
r_ff=r_ff, \
n_resblock=n_resblock,
p_drop=p_drop,
performer_N_opts=performer_N_opts,
performer_L_opts=performer_L_opts,
SE3_param=SE3_param)
self.c6d_predictor = DistanceNetwork(d_pair, p_drop=p_drop)
#
self.refine = Refine_module(n_module_ref, d_node=d_msa, d_pair=130,
d_node_hidden=d_hidden, d_pair_hidden=d_hidden,
SE3_param=REF_param, p_drop=p_drop)
我们可以看到,和之前的pyrosetta模式预测的RoseTTAFoldModule模型类似,同样定义了MSA_emb,Templ_emb,Pair_emb_w_templ,IterativeFeatureExtractor,DistanceNetwork等层次,不同之处在于e2e版本多出了一个部分Refine_module方法。
2.2 Refine_module方法
class Refine_module(nn.Module):
def __init__(self, n_module, d_node=64, d_node_hidden=64, d_pair=128, d_pair_hidden=64,
SE3_param={'l0_in_features':32, 'l0_out_features':16, 'num_edge_features':32}, p_drop=0.0):
super(Refine_module, self).__init__()
self.n_module = n_module
self.proj_edge = nn.Linear(d_pair, d_pair_hidden*2)
self.regen_net = Regen_Network(node_dim_in=d_node, node_dim_hidden=d_node_hidden,
edge_dim_in=d_pair_hidden*2, edge_dim_hidden=d_pair_hidden,
state_dim=SE3_param['l0_out_features'],
nheads=4, nblocks=3, dropout=p_drop)
self.refine_net = _get_clones(Refine_Network(d_node=d_node, d_pair=d_pair_hidden*2,
d_state=SE3_param['l0_out_features'],
SE3_param=SE3_param, p_drop=p_drop), self.n_module)
self.norm_state = LayerNorm(SE3_param['l0_out_features'])
self.pred_lddt = nn.Linear(SE3_param['l0_out_features'], 1)
在该模型中,先是定义了一个全连接层,之后声明了一个自定义的模型Regen_Network再生网络以及一些LayerNorm层,
这个函数可以理解为类型转换函数,将一个不可训练的类型 Tensor 转换成可以训练的类型 parameter 并将这个 parameter 绑定到这个
module 里面(net.parameter() 中就有这个绑定的 parameter,所以在参数优化的时候可以进行优化),所以经过类型转换这个变量就
变成了模型的一部分,成为了模型中根据训练可以改动的参数。使用这个函数的目的也是想让某些变量在学习的过程中不断的修改其
值以达到最优化。
该模型作为一个新的模型被定义,再之后通过_get_clones方法的调用将模型的参数信息进行复制进行持久保存
class Regen_Network(nn.Module):
def __init__(self,
node_dim_in=64,
node_dim_hidden=64,
edge_dim_in=128,
edge_dim_hidden=64,
state_dim=8,
nheads=4,
nblocks=3,
dropout=0.0):
super(Regen_Network, self).__init__()
self.norm_node = LayerNorm(node_dim_in)
self.norm_edge = LayerNorm(edge_dim_in)
self.embed_x = nn.Sequential(nn.Linear(node_dim_in+21, node_dim_hidden), LayerNorm(node_dim_hidden))
self.embed_e = nn.Sequential(nn.Linear(edge_dim_in+2, edge_dim_hidden), LayerNorm(edge_dim_hidden))
blocks = [UniMPBlock(node_dim_hidden,edge_dim_hidden,nheads,dropout) for _ in range(nblocks)]
self.transformer = nn.Sequential(*blocks)
self.get_xyz = nn.Linear(node_dim_hidden,9)
self.norm_state = LayerNorm(node_dim_hidden)
self.get_state = nn.Linear(node_dim_hidden, state_dim)
在Regen_Network中,显式定义了两个LayerNorm层,用于嵌入图信息中点和边的特征,再通过nn.Sequential方法,将全连接层和所说的点、边特征嵌入层连接起来,之后再经过一个自定义的graph transformer模型UniMPBlock,将结果输出。
2.3 MSA方法拓展
两种预测模型在最外层都使用了多重序列比对信息MSA模型,
class MSA_emb(nn.Module):
def __init__(self, d_model=64, d_msa=21, p_drop=0.1, max_len=5000):
super(MSA_emb, self).__init__()
self.emb = nn.Embedding(d_msa, d_model)
self.pos = PositionalEncoding(d_model, p_drop=p_drop, max_len=max_len)
self.pos_q = QueryEncoding(d_model)
def forward(self, msa, idx):
B, N, L = msa.shape
out = self.emb(msa) # (B, N, L, K//2)
out = self.pos(out, idx) # add positional encoding
return self.pos_q(out) # add query encoding
通过在网上查询到的资料,我们可以发现这他们大多都用到了多重序列比对信息(Multiple sequence alignment,MSA),即通过分析序列中的残基共进化(coevolution)信息来改善蛋白三维结构预测。利用MSA进行蛋白结构建模的典型流程如下:
- 对目标蛋白及相关序列进行多重序列比对,实现这一步的常用工具包括Jackhmmer和HHblits;
- 根据序列比对结果,对残基间的进化相关性进行建模。实现这一步的典型做法是训练马尔可夫随机场(Markov Random Field, MRF),如Gremlin[2];
- 根据共进化信息对蛋白结构进行优化。可行的方法包括:将共进化对作为距离约束引入已有的优化器[3],或将其作为特征引入深度学习模型中[1]。
尽管共进化信息对于蛋白结构预测能够提供显著的帮助,上述基于MSA的流程在实际应用中常常会碰到一些问题。近期,来自Facebook AI research的Rao等人在ICLR2021会议上发表了题为“Transformer protein language models are unsupervised structure learners”的论文[Main],提出了一种利用Transformer来替代MSA,通过端到端(end-to-end)的方式直接预测残基间关联性的方法,名为ESM-1b。相比起基于MSA的流程,ESM-1b有如下优势:
- ESM-1b能够绕开MSA流程里许多繁重的计算,如序列检索、序列匹配、MRF模型的训练等,而仅需一次网络前传(forward pass)就能得到结果;
- ESM-1b具有很好的泛化能力,可以用于任意序列,而不像MSA要求有足够多的同源数据。
ESM-1b的训练流程如图1所示。该模型的基本构架是Transformer,是目前自然语言处理(NLP)领域中的一类常用模型。Transformer 的基本思想是通过学习序列补全的方法来学习隐藏在序列中的结构信息。具体地,我们人为遮住序列中的部分残基,并要求模型根据其他残基来预测这些被遮住的残基类型。为了很好地完成这一项任务,深度学习模型需要从数据中学习残基间关联信息(体现在Transformer输出的attention map上)。最后,结合模型输出的attention map以及一个简单的逻辑斯蒂分类器,ESM-1b就可以实现Contact Map的预测了。
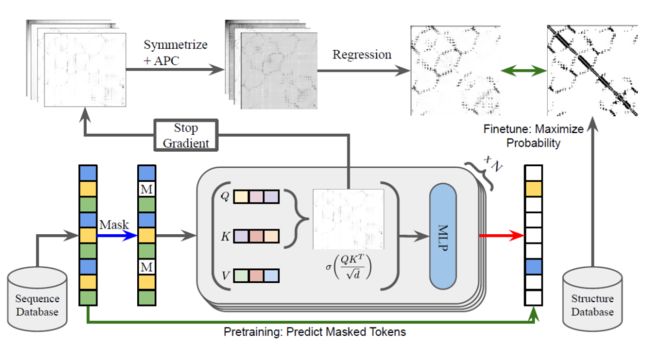
我们注意到在上述流程中,除了最后的逻辑斯蒂分类以外,ESM-1b主体部分(即Transformer)的训练不依赖三维结构信息,仅需未标注的蛋白序列信息,因此该流程在很大程度上是无监督的。ESM-1b模型在无监督预训练时使用了UniRef50数据集[4],在模型评价时选择了trRosetta的训练数据库[5],包含15051个蛋白,经过处理后蛋白数为14882,其中20个数据点用于逻辑斯蒂分类器的训练,其他用于模型评价。
3. 总结
横向比较了一下两种模式的预测,虽然运行e2e的“主攻”是pytorch,需要RoseTTAFold 环境;pyrosetta的“主攻”包括pytorch、tensorflow、pyRosetta,需要RoseTTAFold和folding环境,而且pyRosetta需要在folding环境额外安装,报道中说的也是e2e版本的准确率会相较而言更低,但其实两种模式建立的模型都大同小异,在我看来,区别主要还是体现在了硬件层面和不同的深度学习框架对相关模型的优化上。