(本文于2013.09.04更新)
基因芯片技术的特点是使用寡聚核苷酸探针检测基因。前一节使用ReadAffy函数读取CEL文件获得的数据是探针水平的(probe level),即杂交信号,而芯片数据预处理的目的是将杂交信号转成表达数据(即表达水平数据,expression level data)。存储探针水平数据的是AffyBatch类对象,而表达水平数据为ExpressionSet类对象。基因芯片探针水平数据处理的R软件包有affy, affyPLM, affycomp, gcrma等,这些软件包都很有用。如果没有安装可以通过运行下面R语句安装:
library(BiocInstaller) biocLite(c("affy","gcrma", "affyPLM", "affycomp"))
Affy芯片数据的预处理一般有三个步骤:
- 背景处理(background adjustment)
- 归一化处理(normalization,或称为“标准化处理”)
- 汇总(summarization)。
最后一步获取表达水平数据。需要说明的是,每个步骤都有很多不同的处理方法(算法),选择不同的处理方法对最终结果有非常大的影响。选择哪种方法是仁者见仁智者见智,不同档次的杂志或编辑可能有不同的偏好。
1 需要了解的一点Affy芯片基础知识
Affy基因芯片的探针长度为25个碱基,每个mRNA用11~20个探针去检测,检测同一个mRNA的一组探针称为probe sets。由于探针长度较短,为保证杂交的特异性,affy公司为每个基因设计了两类探针,一类探针的序列与基因完全匹配,称为perfect match(PM)probes,另一类为不匹配的探针,称为mismatch (MM)probes。PM和MM探针序列除第13个碱基外完全一样,在MM中把PM的第13个碱基换成了互补碱基。PM和MM探针成对出现。
我们先使用前一节的方法载入数据并修改芯片名称:
library(affy) library(tcltk) filters <- matrix(c("CEL file", ".[Cc][Ee][Ll]", "All", ".*"), ncol = 2, byrow = T) cel.files <- tk_choose.files(caption = "Select CELs", multi = TRUE, filters = filters, index = 1) # 最好查看一下文件名称 basename(cel.files)
## [1] "NRID9780_Zarka_2-1_MT-0HCA(SOIL)_Rep1_ATH1.CEL" ## [2] "NRID9781_Zarka_2-2_MT-0HCB(SOIL)_Rep2_ATH1.CEL" ## [3] "NRID9782_Zarka_2-3_MT-1HCA(SOIL)_Rep1_ATH1.CEL" ## [4] "NRID9783_Zarka_2-4_MT-1HCB(SOIL)_Rep2_ATH1.CEL" ## [5] "NRID9784_Zarka_2-5_MT-24HCA(SOIL)_Rep1_ATH1.CEL" ## [6] "NRID9785_Zarka_2-6_MT-24HCB(SOIL)_Rep2_ATH1.CEL" ## [7] "NRID9786_Zarka_2-7_MT-7DCA(SOIL)_Rep1_ATH1.CEL" ## [8] "NRID9787_Zarka_2-8_MT-7DCB(SOIL)_Rep2_ATH1.CEL"
data.raw <- ReadAffy(filenames = cel.files) sampleNames(data.raw) <- paste("CHIP",1:length(cel.files),sep="")
用pm和mm函数可查看每个探针的检测情况:
pm.data.raw <- pm(data.raw) head(pm.data.raw, 2)
## CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8 ## 501131 127.0 166.3 112 139.8 111.3 85.5 126.3 102.8 ## 251604 118.5 105.0 82 101.5 94.0 81.3 103.8 103.0
mm.data.raw <- mm(data.raw) head(mm.data.raw, 2)
## CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8 ## 501843 89.0 88.0 80.5 91.0 77.0 75 79.0 72.0 ## 252316 134.3 77.3 77.0 107.8 98.5 75 99.5 71.3
上面显示的列名称就是探针的名称。而基因名称用probeset名称表示:
head(geneNames(data.raw))
## [1] "244901_at" "244902_at" "244903_at" "244904_at" "244905_at" "244906_at"
probeset不一定和实际基因一一对应,这些后面对探针进行基因名称映射时会看到。
2 背景处理(background adjustment)
虽然说是背景处理,但是这一步既处理背景值,又处理噪声信号。注意背景和噪声是两个概念,比如说乡间夜晚的蛙叫声虽嘈杂但很稳定,算是背景,如果突然来一声狗叫,那就是噪声。这两者在统计上可以区分。
芯片的背景处理理论上很简单,因为Affy公司设计MM的目的就是检测非特异杂交信号,PM -MM 不就结了?但是研究发现居然有多达30%的MM探针获得的信号强度比相应PM探针的还强(嘿嘿),所以啊,研究者的饭碗就出来了,整些看起来还合理的方法吧。
R软件包affy用于芯片背景噪声消减的函数是bg.correct(),而MAS和RMA方法是最常用的两种方法。
MAS方法将芯片分为k(默认值为16)个网格区域,用每个区域使用信号强度最低的2%探针去计算背景值和噪声。RMA方法的原理比较复杂,可以参看文献:
R. A. Irizarry, B. Hobbs, F. Collin, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics, 4:249–64, 2003b. 11, 18, 27, 232, 241, 432, 443。
data.rma <- bg.correct(data.raw, method="rma") data.mas <- bg.correct(data.raw, method="mas") class(data.rma)
## [1] "AffyBatch" ## attr(,"package") ## [1] "affy"
class(data.mas)
## [1] "AffyBatch" ## attr(,"package") ## [1] "affy"
class(data.raw)
## [1] "AffyBatch" ## attr(,"package") ## [1] "affy"
可以看到:ReadAffy()读入的CEL芯片数据以AffyBatch类数据形式存储,而背景消减后得到的依然是AffyBatch类数据。
MAS方法应用后PM和MM的信号强度都被重新计算。RMA方法仅使用PM探针数据,背景调整后MM的信号值不变。
head(pm(data.raw)-pm(data.mas),2)
## CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8 ## 501131 79.34 62.93 63.23 72.87 62.48 64.43 62.97 60.86 ## 251604 77.57 63.06 63.73 80.69 63.07 66.53 63.33 60.34
head(pm(data.raw)-pm(data.rma),2)
## CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8 ## 501131 111.2 102.21 93.23 116.36 93.75 76.15 102.82 85.21 ## 251604 103.9 88.69 72.57 90.75 82.23 72.64 87.75 85.32
head(mm(data.raw)-mm(data.mas),2)
## CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8 ## 501843 79.34 62.93 63.24 72.90 62.48 64.44 62.97 60.85 ## 252316 77.56 63.06 63.73 80.66 63.07 66.51 63.33 60.34
#差值为全部为0,说明rma方法没有对mm数值进行处理 head(mm(data.raw)-mm(data.rma),2)
## CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8 ## 501843 0 0 0 0 0 0 0 0 ## 252316 0 0 0 0 0 0 0 0
identical(mm(data.raw), mm(data.rma))
## [1] TRUE
3 归一化处理(normalization)
同一个RNA样品用相同类型的几块芯片进行杂交,获得的结果(信号强度等)都不可能完全相同,甚至差别很大。为了使不同芯片获得的结果具有可比性,必需进行归一化处理。这一步的方法也很多。归一化处理的affy函数为normalize(),以Affybatch对象和处理方法为参数。
3.1 线性缩放方法
这是Affy公司在其软件(4.0和5.0版本)中使用的方法。这种方法先选择一个芯片作为参考,将其他芯片和参考芯片逐一做线性回归分析,用回归直线(没有截距)对其他芯片的信号值做缩放。Affy公司的软件做回归分析前去除了2%最强和最弱信号。使用很简单,mas方法做背景消减后做归一化分析的R语句是:
data.mas.ln <- normalize(data.mas, method = "constant") head(pm(data.mas.ln)/pm(data.mas), 5)
## CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8 ## 501131 1 0.8788 1.002 1.141 0.9929 1.029 0.7578 0.8834 ## 251604 1 0.8788 1.002 1.141 0.9929 1.029 0.7578 0.8834 ## 261891 1 0.8788 1.002 1.141 0.9929 1.029 0.7578 0.8834 ## 230387 1 0.8788 1.002 1.141 0.9929 1.029 0.7578 0.8834 ## 217334 1 0.8788 1.002 1.141 0.9929 1.029 0.7578 0.8834
head(mm(data.mas.ln)/mm(data.mas), 5)
## CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8 ## 501843 1 0.8788 1.002 1.141 0.9929 1.029 0.7578 0.8834 ## 252316 1 0.8788 1.002 1.141 0.9929 1.029 0.7578 0.8834 ## 262603 1 0.8788 1.002 1.141 0.9929 1.029 0.7578 0.8834 ## 231099 1 0.8788 1.002 1.141 0.9929 1.029 0.7578 0.8834 ## 218046 1 0.8788 1.002 1.141 0.9929 1.029 0.7578 0.8834
可以看出,线性缩放方法以第一块芯片为参考,它的数值没有被处理,而其他芯片都被缩放了。对同一块芯片,不同探针的缩放倍数是一个常数。PM和MM的缩放方法完全一样。
3.2 非线性缩放方法
此方法获得的结果比线性方法要好,做非线性拟合时不是取整张芯片而仅取部分(一列)作为基线。
data.mas.nl <- normalize(data.mas, method = "invariantset") head(pm(data.mas.nl)/pm(data.mas), 5)
## CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8 ## 501131 1 1.050 1.417 1.359 1.399 2.023 1.0086 1.3124 ## 251604 1 1.348 2.279 1.838 1.587 2.431 1.1126 1.3059 ## 261891 1 1.564 1.397 1.675 1.497 1.556 1.3167 1.5509 ## 230387 1 1.259 1.683 1.390 1.360 1.504 1.0145 1.2239 ## 217334 1 1.009 1.127 1.241 1.229 1.263 0.8417 0.9934
可以看到,同一芯片不同探针的信号值的缩放倍数是不一样的。
3.3 分位数(quantile)方法
这种方法认为(或假设)每张芯片探针信号的经验分布函数应完全一样,使用任两张芯片的数据做QQ图应该得到一条斜率为1截距为0的直线。
data.mas.qt <- normalize(data.mas, method = "quantiles") head(pm(data.mas.qt)/pm(data.mas), 5)
## CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8 ## 501131 0.7176 0.9602 1.140 1.181 1.112 1.187 0.8145 1.0156 ## 251604 0.6984 0.9814 1.199 1.209 1.112 1.172 0.8287 1.0143 ## 261891 0.6939 0.9956 1.137 1.205 1.111 1.186 0.8389 1.0579 ## 230387 0.7653 0.9777 1.171 1.183 1.115 1.185 0.8153 0.9921 ## 217334 0.9508 0.9547 1.063 1.162 1.130 1.173 0.7945 0.9266
3.4 其他
如循环局部加权回归法(Cyclic loess)和 Contrasts方法。
data.rma.lo <- normalize(data.rma, method = "loess")
## Done with 1 vs 2 in iteration 1 ## Done with 1 vs 3 in iteration 1 ## Done with 1 vs 4 in iteration 1 ## Done with 1 vs 5 in iteration 1 ## Done with 1 vs 6 in iteration 1 ## Done with 1 vs 7 in iteration 1 ## Done with 1 vs 8 in iteration 1 ## Done with 2 vs 3 in iteration 1 ## Done with 2 vs 4 in iteration 1 ## Done with 2 vs 5 in iteration 1 ## Done with 2 vs 6 in iteration 1 ## Done with 2 vs 7 in iteration 1 ## Done with 2 vs 8 in iteration 1 ## Done with 3 vs 4 in iteration 1 ## Done with 3 vs 5 in iteration 1 ## Done with 3 vs 6 in iteration 1 ## Done with 3 vs 7 in iteration 1 ## Done with 3 vs 8 in iteration 1 ## Done with 4 vs 5 in iteration 1 ## Done with 4 vs 6 in iteration 1 ## Done with 4 vs 7 in iteration 1 ## Done with 4 vs 8 in iteration 1 ## Done with 5 vs 6 in iteration 1 ## Done with 5 vs 7 in iteration 1 ## Done with 5 vs 8 in iteration 1 ## Done with 6 vs 7 in iteration 1 ## Done with 6 vs 8 in iteration 1 ## Done with 7 vs 8 in iteration 1 ## 1 0.05775
data.rma.ct <- normalize(data.rma, method = "contrasts")
## Data size 502156 x 8 Desired subset size 5000 +- 50 ## Comuting ranks of old subset....Size of new subset: 109873 ## Comuting ranks of old subset....Size of new subset: 64185 ## Comuting ranks of old subset....Size of new subset: 27795 ## Comuting ranks of old subset....Size of new subset: 14886 ## Comuting ranks of old subset....Size of new subset: 12234 ## Comuting ranks of old subset....Size of new subset: 7835 ## Comuting ranks of old subset....Size of new subset: 7001 ## Comuting ranks of old subset....Size of new subset: 5289 ## Comuting ranks of old subset....Size of new subset: 5230 ## Comuting ranks of old subset....Size of new subset: 5164 ## Comuting ranks of old subset....Size of new subset: 5124 ## Comuting ranks of old subset....Size of new subset: 5065 ## Comuting ranks of old subset....Size of new subset: 5041 ## Fitting normalizing curve ## Normalizing chip 1 ## Normalizing chip 2 ## Normalizing chip 3 ## Normalizing chip 4 ## Normalizing chip 5 ## Normalizing chip 6 ## Normalizing chip 7 ## Normalizing chip 8
4 汇总(Summarization):
最后一步汇总是使用合适的统计方法通过probeset(包含多个探针)的杂交信号计算出计算表达量。affy包的函数为computeExprSet。需要注意的是computeExprSet函数除需要指定统计方法外还需要指定PM校正的方式:computeExprSet(x, pmcorrect.method, summary.method, …)
两个参数可以设定的值可以通过下面函数获得:
pmcorrect.methods()
## [1] "mas" "methods" "pmonly" "subtractmm"
generateExprSet.methods()
## [1] "avgdiff" "liwong" "mas" "medianpolish" ## [5] "playerout"
常用的汇总方法是medianpolish, liwong和mas。liwong方法仅使用PM做背景校正(pmcorrect.method="pmonly")。例如:
eset.rma.liwong <- computeExprSet(data.rma.lo, pmcorrect.method="pmonly", summary.method="liwong")
## 22810 ids to be processed ## | | ## |####################|
计算过程需要一定的时间,耐心等候完成。最后的结果 ExpressionSet 类型数据:
class(eset.rma.liwong)
## [1] "ExpressionSet" ## attr(,"package") ## [1] "Biobase"
class(data.raw)
## [1] "AffyBatch" ## attr(,"package") ## [1] "affy"
5 三合一处理和“自动化”函数:
了解芯片预处理的原理和步骤后你完全可以用一个R函数完成三步处理。affy包提供的三合一处理函数为 expresso(),用法为:
# NOT RUN. 函数说明,不可运行。 expresso( afbatch, bg.correct = TRUE, bgcorrect.method = NULL, bgcorrect.param = list(), normalize = TRUE, normalize.method = NULL, normalize.param = list(), pmcorrect.method = NULL, pmcorrect.param = list(), summary.method = NULL, summary.param = list(), summary.subset = NULL, verbose = TRUE, widget = FALSE)
例如:
# NOT RUN: eset.mas <- expresso(data.raw, bgcorrect.method="mas", normalize.method="constant", pmcorrect.method="mas", summary.method="mas")
使用的各类方法可用bgcorrect.methods,pmcorrect.methods 和 express.summary.stat.methods函数了解。
expresso速度较慢,一些R软件包提供速度较快的预编译三合一函数,如affyPLM软件包的threestep函数。affyPLM提供的处理方法很丰富,有兴趣可以自学。
下面介绍介3个“自动化”函数,这些函数已经预定义了预处理各步骤所采用的方法和参数,不再需要设定。
5.1 mas5函数
由affy包提供:
eset.mas5 <- mas5(data.raw)
## background correction: mas ## PM/MM correction : mas ## expression values: mas ## background correcting...done. ## 22810 ids to be processed ## | | ## |####################|
mas5据说是expresso函数和mas方法的封装。但使用下面语句获得的结果似乎与 mas5(data.raw)的结果不一样(自己去验证看看):
# NOT RUN: expresso(data.raw, bgcorrect.method="mas", normalize=FALSE, pmcorrect.method="mas", summary.method="mas")
5.2 rma函数
也是由affy包提供,其背景处理方法为rma法,归一化处理使用分位数法,而汇总方法使用medianpolish:
eset.rma <- rma(data.raw)
## Background correcting ## Normalizing ## Calculating Expression
它等价于:
# NOT RUN: eset.rma <- expresso(data.raw, bgcorrect.method="rma", normalize.method="quantiles", pmcorrect.method="pmonly", summary.method="medianpolish")
但rma函数是预编译好的C语言函数,速度非常快!
5.3 gcrma函数
由gcrma包提供。 Affy的软件(比如mas方法)使用MM数据做背景处理,但由于MM出现的问题(上面提到过),这些方法可能高估了背景值。而rma方法在做背景处理时没有使用MM数据,这可能又低估了背景值。MM序列公布后有人对其特异性进行了评估,并使用这些评估结果建立了新方法。gcrma就是这样的方法,也是封装好的三合一方法。
library(gcrma) eset.gcrma <- gcrma(data.raw)
## Adjusting for optical effect........Done. ## Computing affinities.Done. ## Adjusting for non-specific binding........Done. ## Normalizing ## Calculating Expression
6 芯片处理方法的优劣评估
芯片预处理的方法这么多,哪个好?我选哪个?知道得越多越迷惑。幸好这些已经有人做了,牛人Rafael Irizarry 和 Leslie Cope专门写了一个R软件包affycomp用于方法评估。
评估方法的优劣必需有数据,而且是包含已知因素的数据。affycomp需要两个系列的数据,一个是RNA稀释系列芯片数据,称为Dilution data,另一个是使用了内参/外标RNA的芯片,称为Spike-in data。Spike-in RNA是在目标物种中不存在、但在芯片上含有相应检测探针的RNA,比如Affy的拟南芥芯片上有几个人或细菌的基因检测探针。由于稀释倍数已知,内参/外标的RNA量和杂交特异性也已知,所以结果可以预测,也就可以用做方法评估。对于严格的芯片实验来说,这些实验都是必须的。但是绝大多数人不做,因为成本很高,尤其是只做几张芯片的时候,一般直接使用别人认可的方法如RMA或MAS。
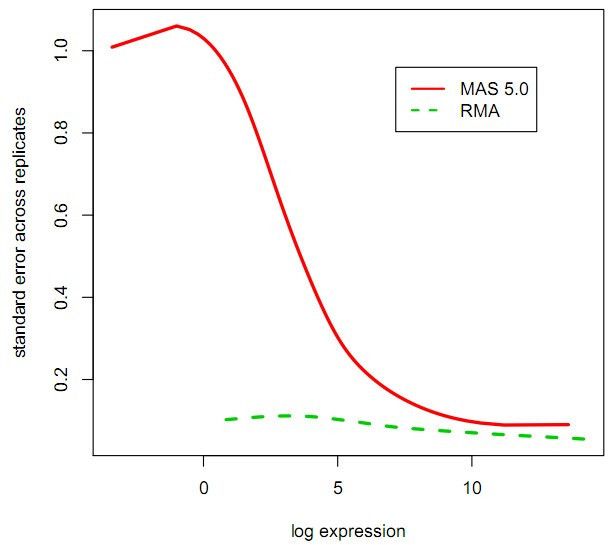
7 Session Info
sessionInfo()
## R version 3.0.1 (2013-05-16) ## Platform: x86_64-pc-linux-gnu (64-bit) ## ## locale: ## [1] LC_CTYPE=zh_CN.UTF-8 LC_NUMERIC=C ## [3] LC_TIME=zh_CN.UTF-8 LC_COLLATE=zh_CN.UTF-8 ## [5] LC_MONETARY=zh_CN.UTF-8 LC_MESSAGES=zh_CN.UTF-8 ## [7] LC_PAPER=C LC_NAME=C ## [9] LC_ADDRESS=C LC_TELEPHONE=C ## [11] LC_MEASUREMENT=zh_CN.UTF-8 LC_IDENTIFICATION=C ## ## attached base packages: ## [1] parallel tcltk stats graphics grDevices utils datasets ## [8] methods base ## ## other attached packages: ## [1] ath1121501probe_2.12.0 gcrma_2.33.1 ath1121501cdf_2.12.0 ## [4] AnnotationDbi_1.23.18 affy_1.39.2 Biobase_2.21.6 ## [7] BiocGenerics_0.7.4 zblog_0.0.1 knitr_1.4.1 ## ## loaded via a namespace (and not attached): ## [1] affyio_1.29.0 BiocInstaller_1.11.4 Biostrings_2.29.15 ## [4] DBI_0.2-7 digest_0.6.3 evaluate_0.4.7 ## [7] formatR_0.9 highr_0.2.1 IRanges_1.19.28 ## [10] preprocessCore_1.23.0 RSQLite_0.11.4 splines_3.0.1 ## [13] stats4_3.0.1 stringr_0.6.2 tools_3.0.1 ## [16] XVector_0.1.0 zlibbioc_1.7.0