分子骨架跃迁工具DiffLinker评测
DiffLinker是一个用于分子骨架跃迁的E3等变3D条件扩散模型。来源于Ilia Igashov等人发表在预印本上,“EQUIVARIANT 3D-CONDITIONAL DIFFUSION MODELS FOR MOLECULAR LINKER DESIGN”文章,链接:https://arxiv.org/pdf/2210.05274.pdf。

与之前介绍的Delinker, 3DLinker等不同,DiffLinker可以连接任意的分子片段,而3Dlinker等仅仅可以链接一对(2个)分子片段。同时,DiffLinker也不需要指定链接处和需要添加的原子数量,这些都可以自动生成。此外,可以使用口袋作为条件,进行骨架跃迁任务。
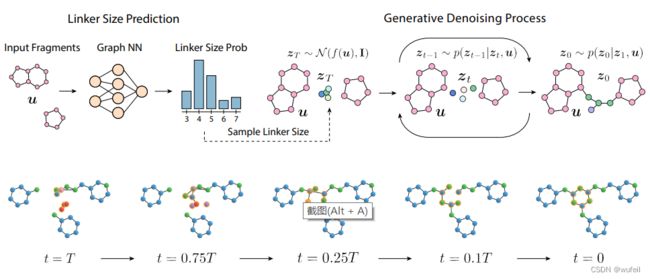
作者提供了github链接:https://github.com/igashov/DiffLinker,并且提供了详细的使用文档。
一、环境安装
复制下载代码:
git clone https://github.com/igashov/DiffLinker.git使用yaml文件安装环境:
conda env create -f environment.yml安装完成后,需要创建models文件夹,并在其中下载一些已经训练好的模型:

这些模型都是作者根据不同的需求训练的模型。
二、linker生成
2.1 测试案例
根据是否使用蛋白口袋的信息,模型可以分为三类:不使用蛋白质口袋(Without protein pocket);仅使用蛋白口袋的骨架(With protein pocket (backbone representation));使用蛋白质口袋的全原子(With protein pocket (full atomic representation))。
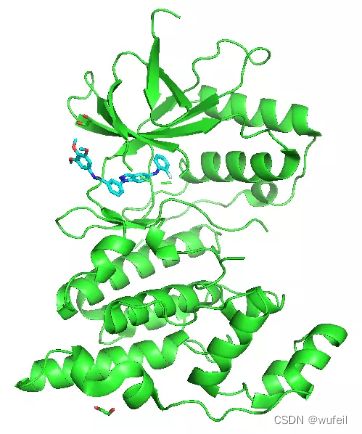
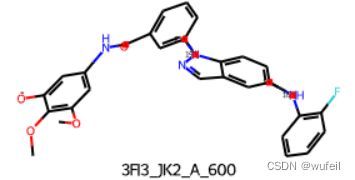
由于作者没有提供示例,所以这里我们自己创建自己的分子骨架跃迁体系。使用之前在3Dlinker中使用过的体系:3FI3及其中小分子,作为例子。
小分子的构象如下图,与DeLinker和3DLinker类似的,以红色框部分,作为骨架跃迁的替换部分。
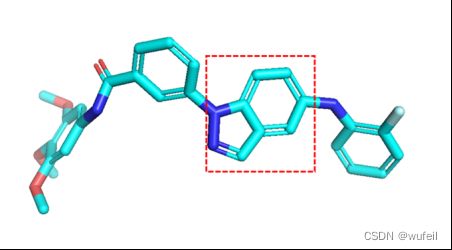
所以,需要进行预处理。
2.2 小分子数据预处理
创建Test_Case文件夹,然后在jupyter notebook中执行如下内容:
导入相关的模块:
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem.Draw import MolDrawing, DrawingOptions
import example_utils
from IPython.display import clear_output
IPythonConsole.ipython_useSVG = True加载原始分子
scaff_1_path = './3FI3_ligand.sdf'
scaff_1_sdf = Chem.SDMolSupplier(scaff_1_path)
scaff_1_smi = Chem.MolToSmiles(scaff_1_sdf[0])
img = Draw.MolsToGridImage([Chem.MolFromSmiles(scaff_1_smi)], molsPerRow=1, subImgSize=(300, 300))
img
对原始分子进行编号,以便进行剪切:
starting_point_3d = Chem.Mol(scaff_1_sdf[0])
example_utils.mol_with_atom_index(starting_point_3d)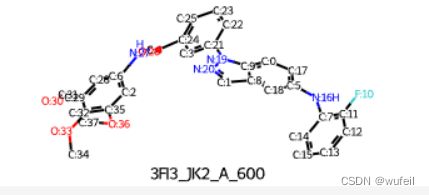
以19和21,5和16号原子组成的单键进行切割。注意骨架跃迁一般仅仅进行非成环的单键的切割,不涉及双键三键等。
atom_pair_idx_1 = [19, 21] atom_pair_idx_2 = [5, 16] bonds_to_break = [starting_point_3d.GetBondBetweenAtoms(x,y).GetIdx() for x,y in [atom_pair_idx_1, atom_pair_idx_2]] fragmented_mol = Chem.FragmentOnBonds(starting_point_3d, bonds_to_break) fragmented_mol
切完以后,生成三个片段,由于未进行坐标更改,所以还是链接在一起的情况。
下面分别进行提取,首先提取左右两侧无需替换的分子片段。
fragments = Chem.GetMolFrags(fragmented_mol,asMols=True)
Draw.MolsToGridImage(fragments[1:], molsPerRow=3) 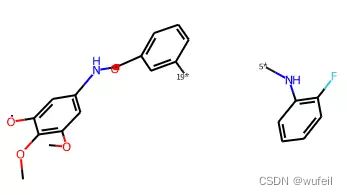
原子进行重新编码:
mol = Chem.CombineMols(fragments[1],fragments[2])
example_utils.mol_with_atom_index(mol) 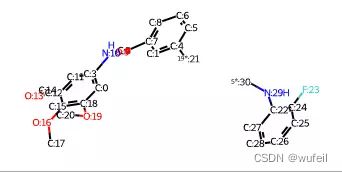
获取断开位置原子对象,并将其原子序号设置为1,即为H原子
atom = mol.GetAtomWithIdx(21) #获取断开位置
atom.SetAtomicNum(1)
atom = mol.GetAtomWithIdx(30)#获取断开位置
atom.SetAtomicNum(1)
mol
将其保存为sdf文件fragments.sdf,
with Chem.SDWriter('fragments.sdf') as w:
w.write(mol) 同理,对骨架跃迁部分的分子片段也进行类似操作
mol2 = fragments[0]
example_utils.mol_with_atom_index(mol2) #分子骨架片段需要重新编码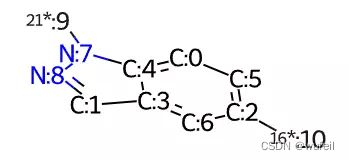
获取断开位置,补氢,保存为fragments_ScanffordHopping.sdf文件。
atom = mol2.GetAtomWithIdx(9) #获取断开位置
atom.SetAtomicNum(1)
atom = mol2.GetAtomWithIdx(10) #获取断开位置
atom.SetAtomicNum(1)
with Chem.SDWriter('fragments_ScanffordHopping.sdf') as w:
w.write(mol2)
同时使用pymol打开两个文件(fragments.sdf和fragments_ScanffordHopping.sdf),断开以后生成的结构(除氢以后)如下:

接下来就可以使用fragments.sdf进行骨架跃迁任务。
2.3 不考虑口袋的骨架跃迁任务
在Test_Case文件夹下创建Without_protein_pocket目录,保存生成的分子。
在Test_Case的上层目录执行如下命令。进行没有口袋限制的分子跃迁任务,生成1000个分子:
python -W ignore generate.py --fragments ./Test_Case/fragments.sdf --model models/geom_difflinker.ckpt --linker_size models/geom_size_gnn.ckpt --output ./Test_Case/Without_protein_pocket --n_samples 1000执行完成后在./Test_Case/Without_protein_pocket目录下会生成1000个sdf文件,分别代表1000个生成的分子。
要注意的是,在文章中,作者提供了多种使用不同数据集训练的模型。我们这里直接使用,文章中结果最好的模型,即使用GEOM数据集训练的结果。
随机打开几个分子,如下:
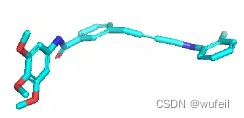
结果说明,DiffLinker生成的分子,有很多错误,没有将两个片段链接一起,或者三键,双键过多。
基于3DLinker和DeLinker类似的分析过程,我们初步测试了生成分子的性能,结果非常不理想:


基于对生成分子的评估,以及生成分子中大量没有将两个片段链接在一起的情况,我们怀疑是输入的fragments.sdf文件存在错误,因为,将dummy原子强制改为H以后,没法通过删除H原子的形式获得smiles。所以,我们尝试在pymol中,强行删除dummy原子,生成新的fragments,即fragments_2.sdf文件,如下:
新建输出文件夹:Without_protein_pocket_2。输出结果仍然不理想,仍然存在大量的断开的现象。感觉相对3Dlinker的生成能力相比,要差了很多。当指定anchor原子为5,29时,结果与之前类似,这里不再赘述。

使用其他的模型,例如:基于ZINC训练出来的,zinc_difflinker.ckpt和zinc_size_gnn.ckpt,结果也是类似:
python -W ignore generate.py \
--fragments ./Test_Case/fragments_2.sdf \
--model models/zinc_difflinker.ckpt \
--linker_size models/zinc_size_gnn.ckpt \
--output ./Test_Case/Without_protein_pocket_4 \
--n_samples 500怀疑时Linker的尺寸,模型估计错误,所以尝试指定特定的linker size,例如:8。
python -W ignore generate.py \
--fragments ./Test_Case/fragments_2.sdf \
--model models/zinc_difflinker.ckpt \
--linker_size 8 \
--output ./Test_Case/Without_protein_pocket_5 \
--n_samples 500结果发现,分子出现断裂的情况,大幅减少。说明,预测linker尺寸的模型存在较为严重的问题,或者对于两个距离较远的fragments的连接原子的预测结果较差。而且,在预测的分子中出现了大量的环。说明linker_size对结果有较为明显的影响。
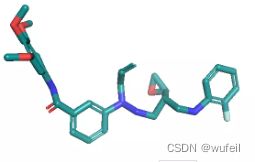
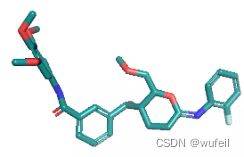
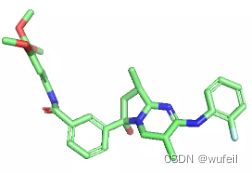

我们也尝试了其他参数,例如: -- n_steps 对结果并没有很多的影响,也许需要更为细致的分析。
2.4 考虑口袋的骨架跃迁任务
需要注意的是,当前使用口袋作为linker生成的条件是无法预测和采样linker的大小的,即,linker size需要指定。同时,需要指定anchor原子。
2.4.1 口袋预处理
使用pymol,对PDBID:3FI3的蛋白进行去除水,然后选择小分子周围6A的原子,保存为pocket.pdb文件,如下图。
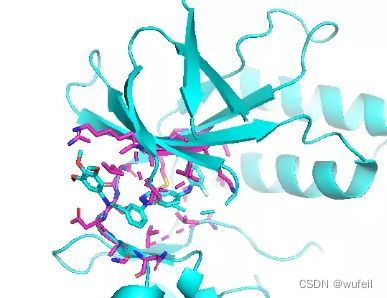
2.4.2 在口袋全原子条件下骨架跃迁
在全口袋全原子限制下的骨架跃迁,生成500个分子,linker的大小定为11个重原子,指定anchor原子为5号和29号原子,输出路径为:./Test_Case/With_protein_pocket_atom, 执行:
python -W ignore generate_with_pocket.py \
--fragments ./Test_Case/fragments_2.sdf \
--pocket ./Test_Case/pocket.pdb \
--model models/pockets_difflinker_full.ckpt \
--output ./Test_Case/With_protein_pocket_atom \
--linker_size 11 \
--anchors 5,29 \
--n_samples 500生成结果保存在./Test_Case/With_protein_pocket_atom文件夹内。
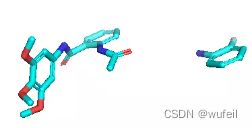
有了口袋的限制,生成的分子质量好了很多,也许也是指定了linker_size和anchor——atom的原因。出现分子断裂的情况少了很多,同时分子大量存在环,特别是双环的情况,说明,模型可能识别到了分子和蛋白的相互作用。
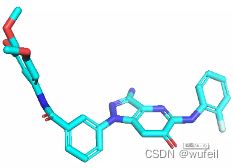

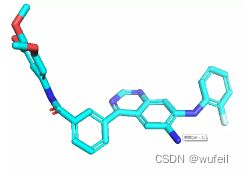

同时,由于口袋条件的存在,生成的分子与口袋发生碰撞的情况也不存在了,这也是生成分子存在环状且为平行口袋空间放置的原因。如下图:蓝色的分子为在口袋条件下的结果,紫色的分子为没有口袋条件的结果。
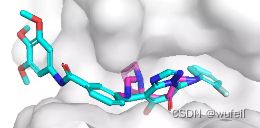
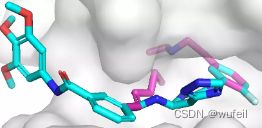
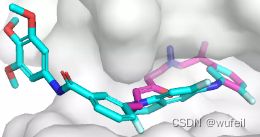

这一结果与文章中的描述一致,文章图如下:

2.4.3 在口袋骨架原子条件下骨架跃迁
使用口袋backbone原子作为条件进行骨架跃迁,执行:
python -W ignore generate_with_pocket.py \
--fragments ./Test_Case/fragments_2.sdf \
--pocket ./Test_Case/pocket.pdb \
--backbone_atoms_only \
--model models/pockets_difflinker_backbone.ckpt \
--output ./Test_Case/With_protein_pocket_backbone \
--linker_size 11 \
--anchors 5,29 \
--n_samples 500运行结束后在/Test_Case/With_protein_pocket_backbon文件夹下会生成500个sdf文件和xyz文件,代表生成的500个分子。
结果与使用口袋全原子作为条件相似,但是,有部分分子与蛋白口袋存在冲突的情况,如下图:

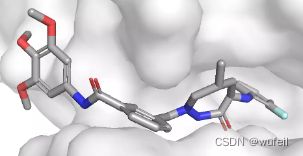
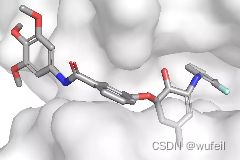
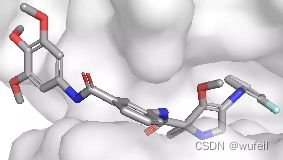
三、DiffLiner生成分子评估
3.1 github评估
GitHub中也提供了评估分子的方法,示例如下:
python -W ignore compute_metrics.py \
ZINC \(dataset)
formatted/zinc_difflinker/sampled_size/zinc_size_gnn/zinc_final_test.smi \(gen_smi_file)
datasets/zinc_final_train_linkers.smi \ (train_set_path)
5 (n_cores) 1(verbose) None(restrict) \
resources/wehi_pains.csv \ (pains_smarts_loc)
diffusion (method)但是该评估方法时针对sample采样出来的分子,如下,但不符合我们的案例情况。
在此之前,还需要将生成的分子进行格式转化。
mkdir -p formatted
python -W ignore reformat_data_obabel.py \
--samples samples \
--dataset zinc_final_test \
--true_smiles_path datasets/zinc_final_test_smiles.smi \
--checkpoint zinc_difflinker \
--formatted formatted \
--linker_size_model_name zinc_size_gnn为此,需要单独改写。
3.2 独立评估
这一部分时根据作者提供的代码进行改写完成。这里以口袋骨架作为生成条件为例,即./With_protein_pocket_backbone文件夹下的分子。
3.2.1 整合生成分子
#合并多个sdf文件,生成
path = './With_protein_pocket_backbone'
files = os.listdir(path)
files = [i for i in files if i.split('.')[-1]=='sdf']
error = []
with Chem.SDWriter(path+'.sdf') as w:
for cid in range(len(files)):
try:
#提取sdf正确的分子,即可以转化为smiles同时可以被smile生成mol对象
with Chem.SDMolSupplier(os.path.join(path, files[cid])) as suppl:
smile = Chem.MolToSmiles(suppl[0])
mol = Chem.MolFromSmiles(smile)
if mol:
w.write(suppl[0])
except:
error.append(files[cid])
print('{} Error!'.format(files[cid]))
print('Len of Error is {}'.format(len(error)))加载原分子
#分子片段
scaff_1_path = './fragments_2.sdf'
scaff_1_sdf = Chem.SDMolSupplier(scaff_1_path)
scaff_1_smi = Chem.MolToSmiles(scaff_1_sdf[0])
img = Draw.MolsToGridImage([Chem.MolFromSmiles(scaff_1_smi)], molsPerRow=1, subImgSize=(300, 300))
img
#完整分子
molecule_1_path = './3FI3_ligand.sdf'
molecule_1_sdf = Chem.SDMolSupplier(molecule_1_path)
molecule_1_smi = Chem.MolToSmiles(molecule_1_sdf[0])
img = Draw.MolsToGridImage([Chem.MolFromSmiles(molecule_1_smi)], molsPerRow=1, subImgSize=(300, 300))
img
分子片段和分子的smiles为:
('COc1cc(NC(=O)c2ccccc2)cc(OC)c1OC.Nc1ccccc1F',
'COc1cc(NC(=O)c2cccc(-n3ncc4cc(Nc5ccccc5F)ccc43)c2)cc(OC)c1OC')
为每一个生成的分子提取Linker
def load_rdkit_molecule(xyz_path, obabel_path, true_frag_smi):
if not os.path.exists(obabel_path):
subprocess.run(f'obabel {xyz_path} -O {obabel_path}', shell=True)
supp = Chem.SDMolSupplier(obabel_path, sanitize=False)
mol = list(supp)[0]
# Keeping only the biggest connected part
mol_frags = Chem.GetMolFrags(mol, asMols=True, sanitizeFrags=False)
mol_filtered = max(mol_frags, default=mol, key=lambda m: m.GetNumAtoms())
try:
mol_smi = Chem.MolToSmiles(mol_filtered)
except RuntimeError:
mol_smi = Chem.MolToSmiles(mol_filtered, canonical=False)
#生成的分子,由于sdf文件中原子的位置存在问题,所以转化一次
mol_filtered = Chem.MolFromSmiles(mol_smi)
# Retrieving linker
true_frag = Chem.MolFromSmiles(true_frag_smi, sanitize=False)
match = mol_filtered.GetSubstructMatch(true_frag)
if len(match) == 0:
linker_smi = ''
else:
elinker = Chem.EditableMol(mol_filtered)
for atom in sorted(match, reverse=True):
elinker.RemoveAtom(atom)
linker = elinker.GetMol()
Chem.Kekulize(linker, clearAromaticFlags=True)
try:
linker_smi = Chem.MolToSmiles(linker)
except RuntimeError:
linker_smi = Chem.MolToSmiles(linker, canonical=False)
return mol_filtered, mol_smi, linker_smi测试一下函数:
mol_filtered, mol_smi, linker_smi = load_rdkit_molecule('./With_protein_pocket_atom/output_304_fragments_2_.xyz', 'output_304_fragments_2_obabel.sdf', scaff_1_smi)
mol_filtered, mol_smi, linker_smi合并所有生成的分子及其骨架:
molecules_sdf = os.listdir('./With_protein_pocket_atom')
molecules_sdf = [m for m in molecules_sdf if m[-3:]=='xyz' and 'obabel' not in m]
data = []
for sdf in molecules_sdf:
try:
in_sdf = os.path.join(path, sdf)
out_sdf = os.path.join(path, sdf[:-4]+'obabel.sdf')
_, mol_smi, linker_smi = load_rdkit_molecule(in_sdf, out_sdf, scaff_1_smi)
data.append({
'sdf': out_sdf,
'fragments': scaff_1_smi,
'true_molecule': molecule_1_smi,
'pred_molecule': mol_smi,
'pred_linker': linker_smi,
})
except:
data.append({
'sdf': out_sdf,
'fragments': scaff_1_smi,
'true_molecule': molecule_1_smi,
'pred_molecule': '',
'pred_linker': '',
})3.2.2 2D评估
记录所有的结果
summary = {}validity有效性检测
在有效分子中含有fragments才算是有效
def is_valid(pred_mol_smiles, frag_smiles):
pred_mol = Chem.MolFromSmiles(pred_mol_smiles)
frag = Chem.MolFromSmiles(frag_smiles)
if frag is None:
return False
if pred_mol is None:
return False
try:
Chem.SanitizeMol(pred_mol, sanitizeOps=Chem.SanitizeFlags.SANITIZE_PROPERTIES)
except Exception:
return False
if len(pred_mol.GetSubstructMatch(frag)) != frag.GetNumAtoms():
return False
return True
valid_cnt = 0
total_cnt = 0
for obj in data:
valid = is_valid(obj['pred_molecule'], obj['fragments'])
obj['valid'] = valid
valid_cnt += valid
total_cnt += 1
validity = valid_cnt / total_cnt * 100
print(f'Validity: {validity:.3f}%')
summary['validity'] = f'{validity:.3f}%'输出:
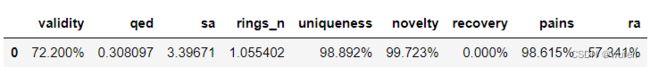
Validity: 72.200%
计算QED
qed_values = []
for obj in data:
if not obj['valid']:
obj['qed'] = None
continue
qed = QED.qed(Chem.MolFromSmiles(obj['pred_molecule']))
obj['qed'] = qed
qed_values.append(qed)
print(f'Mean QED: {np.mean(qed_values):.3f}')
summary['qed'] = np.mean(qed_values)输出:
Mean QED: 0.308
计算SA分数(synthetic accessibility)
sa_values = []
for obj in tqdm(data):
if not obj['valid']:
obj['sa'] = None
continue
#这里对../src/sascorer.py文件进行了修改,增加了_fscores读取路径的参数
sa = sascorer.calculateScore(Chem.MolFromSmiles(obj['pred_molecule']), name='../resources/fpscores')
obj['sa'] = sa
sa_values.append(sa)
print(f'Mean SA: {np.mean(sa_values):.3f}')
summary['sa'] = np.mean(sa_values)输出:
Mean SA: 3.397
计算生成的linker中环的数量
rings_n_values = []
for obj in tqdm(data):
if not obj['valid']:
obj['rings_n'] = None
continue
try:
print(obj['pred_linker'])
rings_n = rdMolDescriptors.CalcNumRings(Chem.MolFromSmiles(obj['pred_linker']))
except:
continue
obj['rings_n'] = rings_n
rings_n_values.append(rings_n)
print(f'Mean Number of Rings: {np.mean(rings_n_values):.3f}')
summary['rings_n'] = np.mean(rings_n_values)输出:
Mean Number of Rings: 1.055
计算分子的独特性(Uniqueness)
true2samples = dict()
for obj in tqdm(data):
if not obj['valid']:
continue
true_mol = obj['true_molecule']
true_frags = obj['fragments']
key = f'{true_mol}_{true_frags}'
true2samples.setdefault(key, []).append(obj['pred_molecule'])
unique_cnt = 0
total_cnt = 0
for samples in tqdm(true2samples.values()):
unique_cnt += len(set(samples))
total_cnt += len(samples)
uniqueness = unique_cnt / total_cnt * 100
print(f'Uniqueness: {uniqueness:.3f}%')
summary['uniqueness'] = f'{uniqueness:.3f}%'输出:
Uniqueness: 98.892%
计算linker的新颖性(Novelty)
这个指标过于苛刻,对于生成的linker在曾经的训练集中也是可以接受的
train_set_path = '/home/ubuntu/wufeil/Deepchem-RDKit/DiffLinker/datasets/zinc_final_train_linkers.smi'
linkers_train = set()
with open(train_set_path, 'r') as f:
for line in f:
linkers_train.add(line.strip())
novel_cnt = 0
total_cnt = 0
for obj in tqdm(data):
if not obj['valid']:
obj['pred_linker_clean'] = None
obj['novel'] = False
continue
try:
linker = Chem.RemoveStereochemistry(obj['pred_linker'])
linker = MolStandardize.canonicalize_tautomer_smiles(Chem.MolToSmiles(linker))
except Exception:
linker = obj['pred_linker']
novel = linker not in linkers_train
obj['pred_linker_clean'] = linker
obj['novel'] = novel
novel_cnt += novel
total_cnt += 1
novelty = novel_cnt / total_cnt * 100
print(f'Novelty: {novelty:.3f}%')
summary['novelty'] = f'{novelty:.3f}%'输出:
Novelty: 99.723%
计算重现率
(Recovery,生成的分子是否可以包含ground true的分子,即真实分子),无效分子不计算在内:
recovered_inputs = set()
all_inputs = set()
for obj in tqdm(data):
if not obj['valid']:
obj['recovered'] = False
continue
key = obj['true_molecule'] + '_' + obj['fragments']
try:
true_mol = Chem.MolFromSmiles(obj['true_molecule'])
Chem.RemoveStereochemistry(true_mol)
true_mol_smi = Chem.MolToSmiles(Chem.RemoveHs(true_mol))
except:
true_mol = Chem.MolFromSmiles(obj['true_molecule'], sanitize=False)
Chem.RemoveStereochemistry(true_mol)
true_mol_smi = Chem.MolToSmiles(Chem.RemoveHs(true_mol, sanitize=False))
pred_mol = Chem.MolFromSmiles(obj['pred_molecule'])
Chem.RemoveStereochemistry(pred_mol)
pred_mol_smi = Chem.MolToSmiles(Chem.RemoveHs(pred_mol))
recovered = true_mol_smi == pred_mol_smi
obj['recovered'] = recovered
if recovered:
recovered_inputs.add(key)
all_inputs.add(key)
recovery = len(recovered_inputs) / len(all_inputs) * 100
print(f'Recovery: {recovery:.3f}%')
summary['recovery'] = f'{recovery:.3f}%'输出:
Recovery: 0.000%
好吧,原分子未出现在生成的分子中。(其实这很有可能是因为分子smiles不同的原因)
PAINS 过滤
泛分析干扰化合物 (PAINS) 检查分子是否不包含在高通量筛选中经常产生假阳性结果的化合物
def check_pains(mol, pains):
for pain in pains:
if mol.HasSubstructMatch(pain):
return False
return True
pains_smarts_loc = '../resources/wehi_pains.csv'
with open(pains_smarts_loc, 'r') as f:
pains_smarts = [Chem.MolFromSmarts(line[0], mergeHs=True) for line in csv.reader(f)]
pains_smarts = set(pains_smarts)
passed_pains_cnt = 0
total_cnt = 0
for obj in tqdm(data):
if not obj['valid']:
obj['passed_pains'] = False
continue
pred_mol = Chem.MolFromSmiles(obj['pred_molecule'])
passed_pains = check_pains(pred_mol, pains_smarts)
obj['passed_pains'] = passed_pains
passed_pains_cnt += passed_pains
total_cnt += 1
pains_score = passed_pains_cnt / total_cnt * 100
print(f'Passed PAINS: {pains_score:.3f}%')
summary['pains'] = f'{pains_score:.3f}%'输出:
Passed PAINS: 98.615%
RA Filter
控制环中共价键顺序的正确性,环的正确性。
def check_ring_filter(linker):
check = True
ssr = Chem.GetSymmSSSR(linker)
for ring in ssr:
for atom_idx in ring:
for bond in linker.GetAtomWithIdx(atom_idx).GetBonds():
if bond.GetBondType() == 2 and bond.GetBeginAtomIdx() in ring and bond.GetEndAtomIdx() in ring:
check = False
return check
passed_ring_filter_cnt = 0
total_cnt = 0
for obj in tqdm(data):
if not obj['valid']:
obj['passed_ring_filter'] = False
continue
pred_linker = Chem.MolFromSmiles(obj['pred_linker'], sanitize=False)
try:
passed_ring_filter = check_ring_filter(pred_linker)
except:
obj['passed_ring_filter'] = False
continue
obj['passed_ring_filter'] = passed_ring_filter
passed_ring_filter_cnt += passed_ring_filter
total_cnt += 1
ra_score = passed_ring_filter_cnt / total_cnt * 100
print(f'Passed Ring Filter: {ra_score:.3f}%')
summary['ra'] = f'{ra_score:.3f}%'输出:
Passed Ring Filter: 57.341%
保存2D分析结果
summary_path = path + '_summary.csv'
summary_table = pd.DataFrame([summary])
summary_table.to_csv(summary_path, index=False)
summary_table输出:
将有效的生成分子保存起来,用于3D分析:
sdf_files = []
data_ = []
for obj in data:
#录入条件,这个可以修改
if obj['passed_ring_filter'] and obj['valid']:
sdf_files.append(obj['sdf'])
data_.append(obj)
with Chem.SDWriter(path+'_valid.sdf') as w:
for cid in range(len(data_)):
with Chem.SDMolSupplier(data_[cid]['sdf']) as suppl:
smile = Chem.MolToSmiles(suppl[0])
mol = Chem.MolFromSmiles(smile)
w.write(suppl[0]) 3.2.3 3D评估
加载原先分子:
true_mol_path = 'fragments_2.sdf'
true_mol_path = '3FI3_ligand.sdf'
true_mol_3d = Chem.SDMolSupplier(true_mol_path)[0]
true_smi2mol3d = {molecule_1_smi: true_mol_3d}
true_smi2mol3d输出:
{'COc1cc(NC(=O)c2cccc(-n3ncc4cc(Nc5ccccc5F)ccc43)c2)cc(OC)c1OC': }
计算SCRDKit相似性
def calc_sc_rdkit_full_mol(gen_mol, ref_mol):
try:
_ = rdMolAlign.GetO3A(gen_mol, ref_mol).Align()
sc_score = calc_SC_RDKit.calc_SC_RDKit_score(gen_mol, ref_mol)
return sc_score
except:
return -0.5
sc_rdkit_list = []
for i, (obj, pred) in tqdm(enumerate(zip(data_, pred_mol_3d)), total=len(data_)):
obj['sc_rdkit'] = None
if not obj['valid']:
continue
true = true_smi2mol3d[obj['true_molecule']]
score = calc_sc_rdkit_full_mol(pred, true)
sc_rdkit_list.append(score)
obj['sc_rdkit'] = score
sc_rdkit_list = np.array(sc_rdkit_list)
sc_rdkit_7 = (sc_rdkit_list > 0.7).sum() / len(sc_rdkit_list) * 100
sc_rdkit_8 = (sc_rdkit_list > 0.8).sum() / len(sc_rdkit_list) * 100
sc_rdkit_9 = (sc_rdkit_list > 0.9).sum() / len(sc_rdkit_list) * 100
sc_rdkit_mean = np.mean(sc_rdkit_list)
print(f'SC_RDKit > 0.7: {sc_rdkit_7:3f}%')
print(f'SC_RDKit > 0.8: {sc_rdkit_8:3f}%')
print(f'SC_RDKit > 0.9: {sc_rdkit_9:3f}%')
print(f'Mean SC_RDKit: {sc_rdkit_mean}')
summary['sc_rdkit_7'] = sc_rdkit_7
summary['sc_rdkit_8'] = sc_rdkit_8
summary['sc_rdkit_9'] = sc_rdkit_9
summary['sc_rdkit_mean'] = sc_rdkit_mean输出:
SC_RDKit > 0.7: 98.067633% SC_RDKit > 0.8: 57.971014% SC_RDKit > 0.9: 0.966184% Mean SC_RDKit: 0.78683985981724
计算RMSD
这一部分计算很有问题,所有分子均计算不出RMSD。后面发现是分子格式的问题,计算分子之间RMSD的时候使用方法不对。修改成如下代码:
def find_exit(mol, num_frag):
neighbors = []
for atom_idx in range(num_frag, mol.GetNumAtoms()):
N = mol.GetAtoms()[atom_idx].GetNeighbors()
for n in N:
if n.GetIdx() < num_frag:
neighbors.append(n.GetIdx())
return neighbors
rmsd_list = []
for i, (obj, pred) in tqdm(enumerate(zip(data, pred_mol_3d)), total=len(data)):
obj['rmsd'] = None
true = true_smi2mol3d[obj['true_molecule']]
Chem.RemoveStereochemistry(true)
true = Chem.RemoveHs(true)
Chem.RemoveStereochemistry(pred)
pred = Chem.RemoveHs(pred)
#由于生成的分子可能存在位置偏差,所以需要先用力场优化一下
AllChem.MMFFOptimizeMolecule(pred)
G1 = frag_utils.topology_from_rdkit(pred)
G2 = frag_utils.topology_from_rdkit(true)
GM = isomorphism.GraphMatcher(G1, G2)
flag = GM.is_isomorphic()
frag_size = Chem.MolFromSmiles(obj['fragments']).GetNumAtoms()
try:
pyO3A = rdMolAlign.GetO3A(pred, true).Matches()
pyO3A = [[(i[0], i[1]) for i in pyO3A[:10]]]
error = rdMolAlign.GetBestRMS(prbMol=pred, refMol=true, map=pyO3A)
num_linker = pred.GetNumAtoms() - frag_size
num_atoms = pred.GetNumAtoms()
error *= np.sqrt(num_atoms / num_linker) # only count rmsd on linker
obj['rmsd'] = error
rmsd_list.append(error)
except:
obj['rmsd'] = None
rmsd_score = np.mean(rmsd_list)
print(f'Mean RMSD: {rmsd_score:.3f}')
summary['rmsd'] = f'{rmsd_score:.3f}'
rmsd_list = np.array([i for i in rmsd_list if i])
rmsd_3 = (rmsd_list < 0.3).sum() / len(rmsd_list) * 100
rmsd_5 = (rmsd_list < 0.5).sum() / len(rmsd_list) * 100
rmsd_7 = (rmsd_list < 0.7).sum() / len(rmsd_list) * 100
rmsd_mean = np.mean(rmsd_list)
print(f'rmsd < 0.3 A: {rmsd_3:3f}%')
print(f'rmsd < 0.5 A: {rmsd_5:3f}%')
print(f'rmsd < 0.7 A: {rmsd_7:3f}%')
print(f'Mean rmsd: {rmsd_mean:4f} A')
summary['rmsd_3'] = rmsd_3
summary['rmsd_5'] = rmsd_5
summary['rmsd_7'] = rmsd_7
summary['rmsd_mean'] = rmsd_mean输出:
Mean RMSD: 0.485 rmsd < 0.3 A: 9.359606% rmsd < 0.5 A: 51.724138% rmsd < 0.7 A: 97.044335% Mean rmsd: 0.484993 A
print(f'Mean rmsd: {rmsd_mean:4f} A')Mean rmsd: 0.484993 A
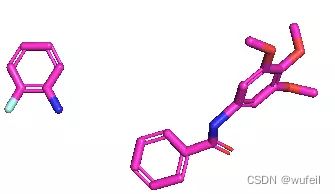
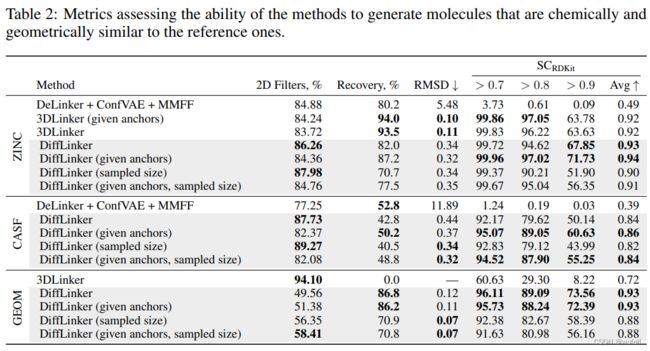
从结果上看,虽然这里仅仅使用一个分子进行了测评,但是整体结果与文章描述大差不差。文章结果如下图:

四、DiffLinker骨架跃迁性能总结
- 对于linker_size的预测DiffLinker做的并不好,所以在使用DiffLinker时需要指定--linker_size参数。指定linker_size参数后,生成分子的准确性大有提高。
- DiffLinker存在明显的分子片段没有被链接的问题,这一点在3DLinker和DeLinker中没有出现。
- 在有口袋条件下的骨架跃迁结果要比没有口袋约束的好,但是仍然存在分子断裂的情况。使用口袋全原子作为条件,预测的分子与口袋发生碰撞的可能小,与文章中描述一致。
3DLinker骨架跃迁生成分子的结果:
3DLinker_generated_molecules
linker_size未指定时DiffLinker骨架跃迁生成分子结果:
DiffLinker_generated_molecules
指定linker_size时DiffLinker骨架跃迁生成分子结果:
DiffLinker_with_LinkerSize
使用口袋全原子作为生成条件时,DiffLinker骨架跃迁生成分子结果:
DiffLinker_with_pocket_atom
使用口袋backbone作为生成条件时,DiffLinker骨架跃迁生成分子结果:
DiffLinker_with_pock_backbone