- 系统学习Python——并发模型和异步编程:进程、线程和GIL
分类目录:《系统学习Python》总目录在文章《并发模型和异步编程:基础知识》我们简单介绍了Python中的进程、线程和协程。本文就着重介绍Python中的进程、线程和GIL的关系。Python解释器的每个实例都是一个进程。使用multiprocessing或concurrent.futures库可以启动额外的Python进程。Python的subprocess库用于启动运行外部程序(不管使用何种
- NGS测序基础梳理01-文库构建(Library Preparation)
qq_21478261
#生物信息生物学
本文介绍Illumina测序平台文库构建(LibraryPreparation)步骤,文库结构。写作时间:2020.05。推荐阅读:10W字《Python可视化教程1.0》来了!一份由公众号「pythonic生物人」精心制作的PythonMatplotlib可视化系统教程,105页PDFhttps://mp.weixin.qq.com/s/QaSmucuVsS_DR-klfpE3-Q10W字《Rg
- NGS测序基础梳理02-簇生成(Cluster Generation)及flow cell介绍
qq_21478261
#生物信息生物信息学
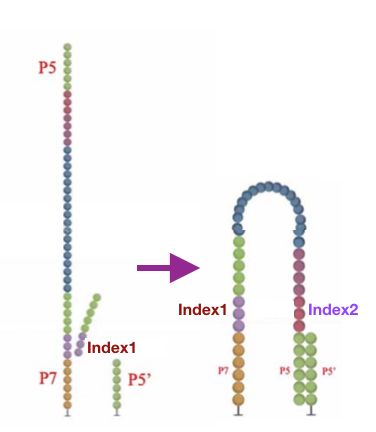
本文图解Illumina测序平台,flowcell表面簇生成(ClusterGeneration)过程。写作时间:2020,有问题可留言或者我的公众号。本文将了解到什么?1flowcell2簇生成为何要进行簇生成?簇生成步骤1)文库与flowcell表面P5杂交与互补链合成2)双链变性3)桥式PCR扩增4)反链切除5)DNA链3'封闭参考资料:1flowcell为何要先介绍flowcell?因为簇
- AWS Terraform 架构指南(二)
绝不原创的飞龙
默认分类默认分类
原文:annas-archive.org/md5/8b2d222956a050c7632b9eee086dadcf译者:飞龙协议:CCBY-NC-SA4.0第七章:7在项目中实现Terraform您准备好开始使用Terraform开发您的AWS基础设施了吗?在本章中,您将学习Terraform的基础知识,并了解如何在AWS中部署您的第一个模板。我们将介绍选择合适的AWS提供商和选择满足您项目需求的
- 电脑选购的基础知识
hello-hebin
有点杂的笔记电脑
文章目录餐前准备电脑的组成电脑选购餐前准备在选购电脑之前先学习一些电脑的基本知识,即电脑的硬件组成,如果你想diy一台比较便宜的高性能的,或者暂时学习了解一些市场的价格,建议点击这里,跳转太平洋电脑城,那么接下来就开始我们的旅途吧!电脑的组成都知道电脑是由硬件和软件组成的,其中硬件基本决定了我们的电脑性能,所有我们在选购电脑时,更加注重的是对硬件的要求,软件的要求并不高,因为软件基本差不多,而且可
- 小学计算机基础知识汇总,电脑基础知识:内存条知识大全,看完小学生都了解...
一、基础知识1、定义、作用内存条又叫随机存取存储器,是一种存储技术,但是和硬盘存储不同,内存条一断电,那么所有数据都会丢失。由于CPU处理器速度很快,而硬盘读写速度完全跟不上CPU的速度,即使是固态硬盘也一样,所以一个急着用,一个慢吞吞,因此就需要一个中间者来帮忙,这就是内存条,硬盘中的数据可以先传输到内存条保存着,如果CPU需要,那么可以直接从内存条中快速读取,相反的,CPU快速处理完后,先放到
- 主板基础知识
bcbobo21cn
硬件主板
主板,又叫主机板(mainboard)、系统板(systemboard)、或母板(motherboard),是计算机最基本的同时也是最重要的部件之一。主板一般为矩形电路板,上面安装了组成计算机的主要电路系统,一般有BIOS芯片、I/O控制芯片、键盘和面板控制开关接口、指示灯插接件、扩充插槽、主板及插卡的直流电源供电接插件等元件。主板制造质量的高低,决定了硬件系统的稳定性。主板与CPU关系密切,每一
- 【电脑】主板的基础知识
Mike_Wuzy
电脑
主板(Motherboard)是计算机的核心组件之一,它将所有其他硬件部件连接在一起并协调它们的工作。以下是关于主板的详细知识:1.架构组成一个典型的主板通常由以下几个主要部分构成:芯片组(Chipset):分为南桥和北桥两个部分。北桥(Northbridge):负责处理高速数据传输,如连接内存控制器、显示接口等。现代CPU集成了北桥的功能,因此许多主板上已经不再有独立的北桥芯片。南桥(South
- Python爬虫实战:基于最新技术的定时签到系统开发全解析
Python爬虫项目
2025年爬虫实战项目python爬虫开发语言人工智能自动化知识图谱
摘要本文详细介绍了如何使用Python开发一个功能完善的定时签到爬虫系统。文章从爬虫基础知识讲起,逐步深入到高级技巧,包括异步请求处理、浏览器自动化、验证码破解、分布式架构等最新技术。我们将通过一个完整的定时签到项目案例,展示如何构建一个稳定、高效且具有良好扩展性的爬虫系统。文中提供了大量可运行的代码示例,涵盖requests、aiohttp、selenium、playwright等多种技术方案,
- 【电脑】CPU的基础知识
Mike_Wuzy
电脑
中央处理器(CentralProcessingUnit,CPU)是计算机的核心部件之一,负责执行程序中的指令并进行计算操作。以下是关于CPU的详细知识:1.架构组成一个典型的现代CPU通常由以下几个主要部分构成:控制单元(ControlUnit,CU):负责从内存中读取指令,并解析这些指令以确定计算机需要完成的操作。算术逻辑单元(ArithmeticLogicUnit,ALU):执行算术运算和逻辑
- 一文读懂HarmonyOS知识地图,开启鸿蒙开发新征程
大雨淅淅
#HarmonyOS开发harmonyos华为
目录一、HarmonyOS知识地图是什么?二、HarmonyOS基础概念速览(一)起源与发展(二)核心特性(三)技术架构剖析1.内核层2.系统服务层3.框架层4.应用层三、HarmonyOS知识地图板块解读(一)开发基础知识1.应用程序包2.应用配置文件3.资源分类与访问4.ArkTS语言基础(二)UI开发知识1.方舟开发框架(ArkUI)2.布局与组件3.动画与交互(三)应用模型与能力1.Abi
- JVM初学者指南:Java虚拟机基础知识 笔记
lenyan~
笔记技术JVMjvmjava笔记
JVM初学者指南:Java虚拟机基础知识全解析摘要:本文记录了Java虚拟机(JVM)的基本概念、架构、内存模型及工作原理的相关笔记-lenyan。一、JVM简介1.1什么是JVM?JVM(JavaVirtualMachine,Java虚拟机)是运行Java字节码的虚拟机。JVM是Java"一次编写,到处运行"这一特性的关键所在。无论什么平台,只要安装了对应的JVM,就能运行Java程序。JVM有
- 篇二 OSI七层模型,TCP/IP四层模型,路由器与交换机原理
苏州向日葵
嵌入式网络开发tcp/ip网络协议网络
一前言本章节主要介绍OSI七层模型,TCP/IP四层模型划分,以及日常使用的路由器,交换机的一些基础知识二OSI七层OSI(OpenSystemsInterconnectionModel)即开放式系统互联模型,是国际标准化组织提出的,一个试图使各种计算机在世界范围内互联为网络的标准框架。层级描述应用层7这一层协议可以理解为面向用户操作行为,无关具体传输,eg:HTTP:浏览网页FTP:文件传输Te
- Python 3.9.0 64位:完整安装与配置教程
D哥有个初二君
本文还有配套的精品资源,点击获取简介:Python3.9.064位安装包为Windows系统上的Python最新版本,特别适用于数据处理、Web开发及自动化脚本等领域。本教程介绍了如何在HarmonyOS开发环境中安装并配置Python3.9.064位版本,包括系统兼容性、下载安装、环境变量配置、安装验证及pip更新。同时提供了Python基础知识,如基础语法、模块导入、面向对象编程、异常处理和文
- 单稳态触发器Multisim电路仿真——硬件工程师笔记
逼子歌
单片机语音识别嵌入式硬件硬件工程师真题硬件工程师硬件工程触发器
目录1单稳态触发器基础知识1.1工作原理1.2电路结构1.3特点1.4应用1.5设计考虑1.6总结2555定时器实现的单稳态触发器2.1电路配置2.2工作原理2.3特点2.4应用2.5设计考虑2.6总结3反相器和与非门实现积分型单稳态触发器3.1电路结构3.2工作原理3.3特点3.4应用3.5设计考虑3.6总结4反相器和与非门实现微分型单稳态触发器4.1电路结构4.2工作原理4.3特点4.4应用4
- 【Java核心计算 基础知识(第9版)】第4章 对象与类
weixin_30872337
java数据结构与算法
本章要点-面向对象程序设计-使用预定义类-用户自定义类-静态域与静态方法-方法参数-对象构造-包-类路径-文档注释-类设计技巧4.1面向对象程序设计概述面向对象的程序是由对象组成的,每个对象包含对用户公开的特定功能部分和隐藏的实现部分。面向过程:算法+数据结构=程序面向对象:数据结构+算法=程序4.1.1类类(class)是构造对象的模板或蓝图。由类构造(construct)对象的过程称为创建类的
- Python基础知识4
QQLOVEYY
Python学习pythonpycharm
复习自学自用,不适合全面学习的家人们,想看的可以看一下一、标准库与第三方库标准库是Python自带的“宝藏库”,涵盖了众多实用功能。其中包括内置函数,像我们常用的print用于输出信息、input用于获取用户输入;还有内置类型,如int(整数)、str(字符串)、bool(布尔值)、list(列表)、dict(字典)等,它们是构建Python程序的基础数据结构。此外,标准库还涉及文本处理、时间日期
- Python 基础知识1
QQLOVEYY
Python学习pythonpycharm
只是用来自学python并复习的,如果想看可以看一下,不建议全面学习的看一、基本输出与字面值常量在Python中,print()函数是实现输出功能的基础工具。它既可以输出字符串,也能直接对算术表达式进行计算并输出结果。示例代码:print('hello')#输出字符串'hello'print(1+2-3)#输出0,执行1+2-3的算术运算print(1+2*3)#输出7,遵循先乘除后加减的运算优先
- Python基础知识2
QQLOVEYY
Python学习pythonpycharm
二、顺序语句:程序执行的基础路径2.1执行原理顺序语句是Python程序最基础的执行模式,代码按照编写顺序,从上至下、逐行执行,每条语句仅执行一次,直至程序结束或遇到控制流语句改变执行方向。2.2示例代码print("第一步操作")print("第二步操作")print("第三步操作")执行结果:第一步操作第二步操作第三步操作三、条件语句:基于条件的决策执行3.1if-else结构3.1.1语法规
- FreeRTOS基础知识学习指南
以下内容涵盖FreeRTOS的核心概念,包括任务管理、调度、中断、互斥量与信号量、队列和内存管理等主题。每部分提供基本原理说明,并辅以简要的代码示例帮助理解。1.任务管理(TaskManagement)任务的创建与删除:FreeRTOS中的任务相当于独立的线程。可以使用xTaskCreate()动态创建任务,或使用xTaskCreateStatic()静态创建任务(提供预先分配的栈和控制块内存)。
- 人工智能基础知识PPT课件
智慧化智能化数字化方案
方案解读馆人工智能入门人工智能学习人工智能课件人工智能PPT
人工智能基础知识定义与概念:人工智能是研究、开发用于模拟、延伸和扩展人类智能行为的综合性科学,其目的是让计算机系统具备执行人类智能任务的能力。涉及计算机科学、数学等多学科,研究对象是让系统具备智能,智能包括认知、适应和自主能力等维度。学派与方法学派:有符号主义、联结主义、行为主义等学派,分别从不同角度研究人工智能。方法:包括基于知识、学习和仿生的方法,如专家系统、机器学习、深度学习等。分类与发展分
- 网络通信与部署基础知识
伤心美眉
互联网网络
网络通信是互联网的支柱,无论是访问网页、传输文件还是搭建服务,都离不开网络。本文将详细讲解网络通信的演变、端口、Socket与TCP、内网与外网部署、防火墙配置、公司服务器与云服务器的关系,以及VPN的原理和使用方法。1.网络通信的演变网络通信从简单到复杂,逐步解决了多设备连接和高效传输的问题。以下是三个阶段的详细讲解。1.1早期:点对点通信知识点:在网络发展的初期(几十年前),通信方式非常简单:
- Redis核心用法与通用命令全解析
Pota-to成长日记
Redisredis数据库缓存
Redis核心用法与通用命令全解析——从基础操作到高效实践一、Redis基础知识速览Redis是一款高性能的键值存储系统,支持String、Hash、List、Set、SortedSet五种核心数据结构,以及Bitmaps、HyperLogLog、Streams等扩展类型。其单线程模型和内存存储特性使其在缓存、计数器、消息队列等场景中表现出色。二、核心命令详解(附实用示例)1.通用键操作命令(1)
- [C语言初阶]指针初阶
目录一、指针是什么?二、指针与指针类型三、野指针及其避免方法3.1什么是野指针?3.2野指针产生的原因:3.3如何避免野指针?四、指针运算4.1应用:实现strlen函数五、指针与数组六、二级指针七、指针数组指针是C语言的灵魂所在,也是许多初学者感到困惑的概念。本文将带你系统学习指针的基础知识,从指针的本质到指针运算,再到指针与数组的关系,最后介绍二级指针和指针数组的概念。通过本文的学习,你将建立
- scanpy保存图片的常用方法汇总
Bio Coder
空间转录组&单细胞scanpy保存图片汇总
在使用Scanpy(一个用于单细胞RNA测序数据分析的Python库)时,保存图片(如可视化结果)是常见的操作。Scanpy的绘图功能主要基于Matplotlib和Seaborn,保存图片的方法也与这些库的保存机制一致。以下是Scanpy保存图片的详细方法及注意事项:1.基本保存图片的方法Scanpy的绘图函数(如sc.pl.umap、sc.pl.tsne、sc.pl.pca等)通常会返回Matp
- 隐马尔可夫模型(HMM):观测背后的状态解码艺术
大千AI助手
人工智能Python#OTHER数据挖掘人工智能机器学习算法HMM马尔科夫概率论
本文由「大千AI助手」原创发布,专注用真话讲AI,回归技术本质。拒绝神话或妖魔化。搜索「大千AI助手」关注我,一起撕掉过度包装,学习真实的AI技术!一、核心概念:双重随机过程隐马尔可夫模型(HiddenMarkovModel,HMM)是一种通过可观测序列推断隐含状态序列的概率图模型,包含两个核心随机过程:隐含状态链:不可观测的马尔可夫过程${q_t}$P(qt∣qt−1,qt−2,…,q1)=P(
- Next.js 开发指南 实战篇 | React Notes | 项目介绍与创建
人工智能_SYBH
课程推荐javascriptreact.js前端开发语言Next.js
Next.js开发指南-冴羽-掘金小册前言欢迎来到实战篇!基础篇的目标是带大家复习基础知识,以及用作使用手册,方便大家在以后的项目开发中查询API用法,属于这本小册的“赠送面积”。从本篇起就进入小册的正式内容了。我们的第一个实战项目是ReactNotes,因为Next.jsv14基于ReactServerComponent构建的AppRouter,而ReactServerComponent的起源是
- React源码2 React中的工厂函数:createRoot()
gzzeason
ReactV18.2源码react.jsjavascript前端
#ReactV18.2源码前置基础知识:工厂函数工厂函数是一种设计模式,用于动态创建对象或函数实例。其核心思想是通过封装对象创建的细节,提供统一的接口,从而增强代码的灵活性和可维护性,有一些核心作用:解耦创建逻辑:将对象的实例化过程与使用分离,调用方无需关心具体实现细节。动态生成:根据输入参数返回不同类型的对象或函数。统一接口:通过单一入口点管理多种创建场景。工厂函数由构造函数进阶而来,都是用来创
- 大唐杯省赛考纲总结(10%)
LUO-CHEn
大唐杯第十届5G信息与通信
系列文章目录本届大唐杯考察范围20%通信基础知识70%5G内容10%商业流程文章目录系列文章目录前言一、通信基础知识的考察(20%)二、5G内容5G无线技术(20%):5G网络技术(10%):5G协议与信令(15%):5G工程实践(15%):5G+垂直行业应用(10%):三、商业流程(10%):总结前言大唐杯以推广信息通信领域前沿技术、协同高校学科建设、推动行业创新发展为目的,激发高校学生参赛热情
- NodeJS全栈WEB3面试题——P1基础知识:区块链与Web3原理
穗余
Web3web3区块链
1.1区块链的基本组成有哪些部分?它们的作用是什么?区块链主要由以下几个部分组成:区块(Block):区块是数据的基本存储单元,每个区块包含一批交易记录和一些元数据(如时间戳、前一个区块的哈希等)。链(Chain):区块通过哈希指针连接形成链条,保证数据的不可篡改性。节点(Node):参与网络的计算机,每个节点维护区块链的完整或部分副本。共识机制(ConsensusMechanism):用于节点间
- 对于规范和实现,你会混淆吗?
yangshangchuan
HotSpot
昨晚和朋友聊天,喝了点咖啡,由于我经常喝茶,很长时间没喝咖啡了,所以失眠了,于是起床读JVM规范,读完后在朋友圈发了一条信息:
JVM Run-Time Data Areas:The Java Virtual Machine defines various run-time data areas that are used during execution of a program. So
- android 网络
百合不是茶
网络
android的网络编程和java的一样没什么好分析的都是一些死的照着写就可以了,所以记录下来 方便查找 , 服务器使用的是TomCat
服务器代码; servlet的使用需要在xml中注册
package servlet;
import java.io.IOException;
import java.util.Arr
- [读书笔记]读法拉第传
comsci
读书笔记
1831年的时候,一年可以赚到1000英镑的人..应该很少的...
要成为一个科学家,没有足够的资金支持,很多实验都无法完成
但是当钱赚够了以后....就不能够一直在商业和市场中徘徊......
- 随机数的产生
沐刃青蛟
随机数
c++中阐述随机数的方法有两种:
一是产生假随机数(不管操作多少次,所产生的数都不会改变)
这类随机数是使用了默认的种子值产生的,所以每次都是一样的。
//默认种子
for (int i = 0; i < 5; i++)
{
cout<<
- PHP检测函数所在的文件名
IT独行者
PHP函数
很简单的功能,用到PHP中的反射机制,具体使用的是ReflectionFunction类,可以获取指定函数所在PHP脚本中的具体位置。 创建引用脚本。
代码:
[php]
view plain
copy
// Filename: functions.php
<?php&nbs
- 银行各系统功能简介
文强chu
金融
银行各系统功能简介 业务系统 核心业务系统 业务功能包括:总账管理、卡系统管理、客户信息管理、额度控管、存款、贷款、资金业务、国际结算、支付结算、对外接口等 清分清算系统 以清算日期为准,将账务类交易、非账务类交易的手续费、代理费、网络服务费等相关费用,按费用类型计算应收、应付金额,经过清算人员确认后上送核心系统完成结算的过程 国际结算系
- Python学习1(pip django 安装以及第一个project)
小桔子
pythondjangopip
最近开始学习python,要安装个pip的工具。听说这个工具很强大,安装了它,在安装第三方工具的话so easy!然后也下载了,按照别人给的教程开始安装,奶奶的怎么也安装不上!
第一步:官方下载pip-1.5.6.tar.gz, https://pypi.python.org/pypi/pip easy!
第二部:解压这个压缩文件,会看到一个setup.p
- php 数组
aichenglong
PHP排序数组循环多维数组
1 php中的创建数组
$product = array('tires','oil','spark');//array()实际上是语言结构而不 是函数
2 如果需要创建一个升序的排列的数字保存在一个数组中,可以使用range()函数来自动创建数组
$numbers=range(1,10)//1 2 3 4 5 6 7 8 9 10
$numbers=range(1,10,
- 安装python2.7
AILIKES
python
安装python2.7
1、下载可从 http://www.python.org/进行下载#wget https://www.python.org/ftp/python/2.7.10/Python-2.7.10.tgz
2、复制解压
#mkdir -p /opt/usr/python
#cp /opt/soft/Python-2
- java异常的处理探讨
百合不是茶
JAVA异常
//java异常
/*
1,了解java 中的异常处理机制,有三种操作
a,声明异常
b,抛出异常
c,捕获异常
2,学会使用try-catch-finally来处理异常
3,学会如何声明异常和抛出异常
4,学会创建自己的异常
*/
//2,学会使用try-catch-finally来处理异常
- getElementsByName实例
bijian1013
element
实例1:
<!DOCTYPE html PUBLIC "-//W3C//DTD XHTML 1.0 Transitional//EN" "http://www.w3.org/TR/xhtml1/DTD/xhtml1-transitional.dtd">
<html xmlns="http://www.w3.org/1999/x
- 探索JUnit4扩展:Runner
bijian1013
java单元测试JUnit
参加敏捷培训时,教练提到Junit4的Runner和Rule,于是特上网查一下,发现很多都讲的太理论,或者是举的例子实在是太牵强。多搜索了几下,搜索到两篇我觉得写的非常好的文章。
文章地址:http://www.blogjava.net/jiangshachina/archive/20
- [MongoDB学习笔记二]MongoDB副本集
bit1129
mongodb
1. 副本集的特性
1)一台主服务器(Primary),多台从服务器(Secondary)
2)Primary挂了之后,从服务器自动完成从它们之中选举一台服务器作为主服务器,继续工作,这就解决了单点故障,因此,在这种情况下,MongoDB集群能够继续工作
3)挂了的主服务器恢复到集群中只能以Secondary服务器的角色加入进来
2
- 【Spark八十一】Hive in the spark assembly
bit1129
assembly
Spark SQL supports most commonly used features of HiveQL. However, different HiveQL statements are executed in different manners:
1. DDL statements (e.g. CREATE TABLE, DROP TABLE, etc.)
- Nginx问题定位之监控进程异常退出
ronin47
nginx在运行过程中是否稳定,是否有异常退出过?这里总结几项平时会用到的小技巧。
1. 在error.log中查看是否有signal项,如果有,看看signal是多少。
比如,这是一个异常退出的情况:
$grep signal error.log
2012/12/24 16:39:56 [alert] 13661#0: worker process 13666 exited on s
- No grammar constraints (DTD or XML schema).....两种解决方法
byalias
xml
方法一:常用方法 关闭XML验证
工具栏:windows => preferences => xml => xml files => validation => Indicate when no grammar is specified:选择Ignore即可。
方法二:(个人推荐)
添加 内容如下
<?xml version=
- Netty源码学习-DefaultChannelPipeline
bylijinnan
netty
package com.ljn.channel;
/**
* ChannelPipeline采用的是Intercepting Filter 模式
* 但由于用到两个双向链表和内部类,这个模式看起来不是那么明显,需要仔细查看调用过程才发现
*
* 下面对ChannelPipeline作一个模拟,只模拟关键代码:
*/
public class Pipeline {
- MYSQL数据库常用备份及恢复语句
chicony
mysql
备份MySQL数据库的命令,可以加选不同的参数选项来实现不同格式的要求。
mysqldump -h主机 -u用户名 -p密码 数据库名 > 文件
备份MySQL数据库为带删除表的格式,能够让该备份覆盖已有数据库而不需要手动删除原有数据库。
mysqldump -–add-drop-table -uusername -ppassword databasename > ba
- 小白谈谈云计算--基于Google三大论文
CrazyMizzz
Google云计算GFS
之前在没有接触到云计算之前,只是对云计算有一点点模糊的概念,觉得这是一个很高大上的东西,似乎离我们大一的还很远。后来有机会上了一节云计算的普及课程吧,并且在之前的一周里拜读了谷歌三大论文。不敢说理解,至少囫囵吞枣啃下了一大堆看不明白的理论。现在就简单聊聊我对于云计算的了解。
我先说说GFS
&n
- hadoop 平衡空间设置方法
daizj
hadoopbalancer
在hdfs-site.xml中增加设置balance的带宽,默认只有1M:
<property>
<name>dfs.balance.bandwidthPerSec</name>
<value>10485760</value>
<description&g
- Eclipse程序员要掌握的常用快捷键
dcj3sjt126com
编程
判断一个人的编程水平,就看他用键盘多,还是鼠标多。用键盘一是为了输入代码(当然了,也包括注释),再有就是熟练使用快捷键。 曾有人在豆瓣评
《卓有成效的程序员》:“人有多大懒,才有多大闲”。之前我整理了一个
程序员图书列表,目的也就是通过读书,让程序员变懒。 程序员作为特殊的群体,有的人可以这么懒,懒到事情都交给机器去做,而有的人又可以那么勤奋,每天都孜孜不倦得
- Android学习之路
dcj3sjt126com
Android学习
转自:http://blog.csdn.net/ryantang03/article/details/6901459
以前有J2EE基础,接触JAVA也有两三年的时间了,上手Android并不困难,思维上稍微转变一下就可以很快适应。以前做的都是WEB项目,现今体验移动终端项目,让我越来越觉得移动互联网应用是未来的主宰。
下面说说我学习Android的感受,我学Android首先是看MARS的视
- java 遍历Map的四种方法
eksliang
javaHashMapjava 遍历Map的四种方法
转载请出自出处:
http://eksliang.iteye.com/blog/2059996
package com.ickes;
import java.util.HashMap;
import java.util.Iterator;
import java.util.Map;
import java.util.Map.Entry;
/**
* 遍历Map的四种方式
- 【精典】数据库相关相关
gengzg
数据库
package C3P0;
import java.sql.Connection;
import java.sql.SQLException;
import java.beans.PropertyVetoException;
import com.mchange.v2.c3p0.ComboPooledDataSource;
public class DBPool{
- 自动补全
huyana_town
自动补全
<!DOCTYPE html PUBLIC "-//W3C//DTD XHTML 1.0 Transitional//EN" "http://www.w3.org/TR/xhtml1/DTD/xhtml1-transitional.dtd"><html xmlns="http://www.w3.org/1999/xhtml&quo
- jquery在线预览PDF文件,打开PDF文件
天梯梦
jquery
最主要的是使用到了一个jquery的插件jquery.media.js,使用这个插件就很容易实现了。
核心代码
<!DOCTYPE html PUBLIC "-//W3C//DTD XHTML 1.0 Transitional//EN" "http://www.w3.org/TR/xhtml1/DTD/xhtml1-transitional.
- ViewPager刷新单个页面的方法
lovelease
androidviewpagertag刷新
使用ViewPager做滑动切换图片的效果时,如果图片是从网络下载的,那么再子线程中下载完图片时我们会使用handler通知UI线程,然后UI线程就可以调用mViewPager.getAdapter().notifyDataSetChanged()进行页面的刷新,但是viewpager不同于listview,你会发现单纯的调用notifyDataSetChanged()并不能刷新页面
- 利用按位取反(~)从复合枚举值里清除枚举值
草料场
enum
以 C# 中的 System.Drawing.FontStyle 为例。
如果需要同时有多种效果,
如:“粗体”和“下划线”的效果,可以用按位或(|)
FontStyle style = FontStyle.Bold | FontStyle.Underline;
如果需要去除 style 里的某一种效果,
- Linux系统新手学习的11点建议
刘星宇
编程工作linux脚本
随着Linux应用的扩展许多朋友开始接触Linux,根据学习Windwos的经验往往有一些茫然的感觉:不知从何处开始学起。这里介绍学习Linux的一些建议。
一、从基础开始:常常有些朋友在Linux论坛问一些问题,不过,其中大多数的问题都是很基础的。例如:为什么我使用一个命令的时候,系统告诉我找不到该目录,我要如何限制使用者的权限等问题,这些问题其实都不是很难的,只要了解了 Linu
- hibernate dao层应用之HibernateDaoSupport二次封装
wangzhezichuan
DAOHibernate
/**
* <p>方法描述:sql语句查询 返回List<Class> </p>
* <p>方法备注: Class 只能是自定义类 </p>
* @param calzz
* @param sql
* @return
* <p>创建人:王川</p>
* <p>创建时间:Jul
 img
img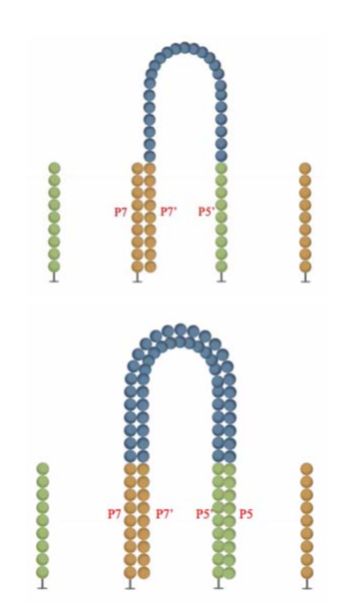 img
img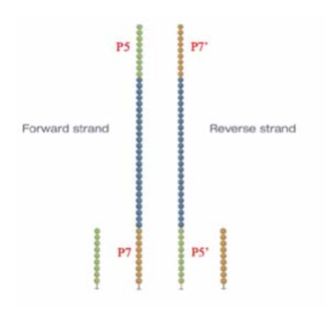 img
img