生信学习笔记:fastp质控处理生成的report结果解读
文章目录
-
- 前言
- raw data 和 fastq文件
- reads
- Q20和Q30
- N值
- Adapters
- Duplication
- Insert
- fastp report
-
- summary
- Adapter
- Insert size estimation
- Before filtering
前言
测序出来的数据利用fastp一个命令质控全搞定,无论是SE还是PE,同时会生成.json和.html格式的报告,十分直观方便,如何生成报告可查看 Linux下fastp的使用 ,下面记录一下如何理解这份报告。
在这之前先整理几个概念:
raw data 和 fastq文件
测序得到的原始图像数据经base calling 转化为序列数据,我们称之为raw data或raw reads,结果以fastq 文件格式存储,fastq文件为用户得到的最原始文件,里面存储 reads的序列以及reads的测序质量。
在fastq 格式文件中每个read由四行描述:
1.@read ID
2.TGGCGGAGGGATTTGAACCC
3.+
4.bbbbbbbbabbbbbbbbbbb
每个序列共有4行,第1行和第3行是序列名称(有的fq文件为了节省存储空间会省略第三行“+"后面的序列名称);
第2行是序列;
第4行是序列的测序质量,每个字符对应第2行每个碱基,第4行每个字符对应的ASClI值减去64,即为该碱基的测序质量值,比如h对应的ASCIl值为104,那么其对应的碱基质量值是40。(碱基质量值范围为0到40)
下表为Solexa 测序错误率与测序质量值简明对应关系:
| 测序错误率 | 测序质量值 | 对应字符 |
|---|---|---|
| 5% | 13 | M |
| 1% | 20 | T |
| 0.1% | 30 | ^ |
| 0.01% | 40 | h |
公式:-10*log10P
fastp就是对.fa.gz格式的文件进行处理
reads
由于受目前测序水平的限制,基因组测序时需要先将基因组打断成DNA片段,然后再建库测序。reads(读长)指的是测序仪单次测序所得到的碱基序列,也就是一连串的ATCGGGTA之类的,它不是基因组中的组成。不同的测序仪器,reads长度不一样。对整个基因组进行测序,就会产生成百上千万的reads。

- 高通量测序时,在芯片上的每个反应,会读出一条序列,是比较短的,叫read,它们是原始数据;
- 有很多reads通过片段重叠,能够组装成一个更大的片段,称为contig;多个contigs通过片段重叠,组成一个更长的scaffold;
- 一个contig被组成出来之后,鉴定发现它是编码蛋白质的基因,就叫singleton;
- 多个contigs组装成scaffold之后,鉴定发现它编码蛋白质的基因,叫unigene.
Q20和Q30
Q20,Q30它们代表的是某一碱基质量值占全部碱基数的百分比,就类似于产品合格率,不同的质量标准会产生不同的合格率,标准越高,质量越好,达标的就越少;合格率越高,那么达标的数据就越多。一般来说,对于二代测序,最好是达到Q20的碱基要在95%以上(最差不低于90%),Q30要求大于85%(最差也不要低于80%)。
一个给定碱基的测序质量分值Q定义为下面的等式:
Q = -10log10(e)
其中,e为预计碱基检出不正确的概率。
Q分值较高表示出错的概率较小。
Q分值较低可能会导致相当大一部分的片段不可用,还可能导致假阳性的变异检出增加,以致得出不准确的结论。
测量分值与碱基检出精度的关系如下:
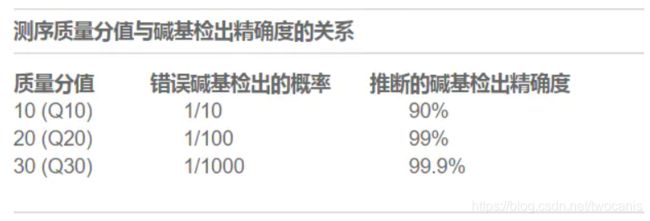
N值
N 代表没有测定的碱基。(ATCG都有可能)比如在测序过程中出现gap,那么这一段都用N来代替这些还没有测序、尚不明确的碱基。
Adapters
adapter
接头,为一段已知的短核苷酸序列,用于链接未知的目标测 序片段
index或barcode
几个碱基组成的寡核苷酸链,用于在混合测序时,区分不同样本
可根据fastq序列中的信息获取
@HWI-ST1276:71:C1162ACXX:1:1101:1208:2458 1:N:0:CGATGT
即第一行最末的 CGATGT 即本次测序所使用的index。
insert
待测序的目标序列,位于两个adapter之间

Duplication
Duplication Rate = 1- Unique reads/Total reads
cluster,是指二代测序所用芯片表面或单个磁珠表面生成的由单个DNA模板生成的数百至数千个DNA分子的集合,犹如单个细菌在LB培养基表面生成单个菌落。
Duplication Reads,是指多个完全相同的DNA片段形成了多个有效cluster,读取这些Cluster所获得reads信息也是完全相同,被称之为Duplication reads
RNAseq与16S去duplication问题
1、RNAseq与16s测序的duplication并不是打断不随机造成的,不能去除duplication
2、去除duplication会造成丰度信息丢失
常见文库的Duplication Rate经验值
WES(全外显子组测序),~10G,dup rate在10%左右;
WGS(全基因组测序),~90G,dup rate在10%左右;
RNA-seq(转录组测序技术),dup rate在40%~50%左右;
WGBS(全基因组甲基化测序),>10G, dup rate > 10%;
多重PCR文库和Panel,差异很大,跟需要测序的区域以及测序量有关,通常情况下只要on target部分数据质量足够好,dup rate不是一个重要的考虑指标。
Insert
插入片段,通俗解释就是两个Adapter接头中间的,被read的片段,即被打断的目标片段
详情可见这篇一篇文章说清楚什么是“插入片段”?,说的很清楚
fastp report
summary
首先是一个总的报告,我处理的是PE
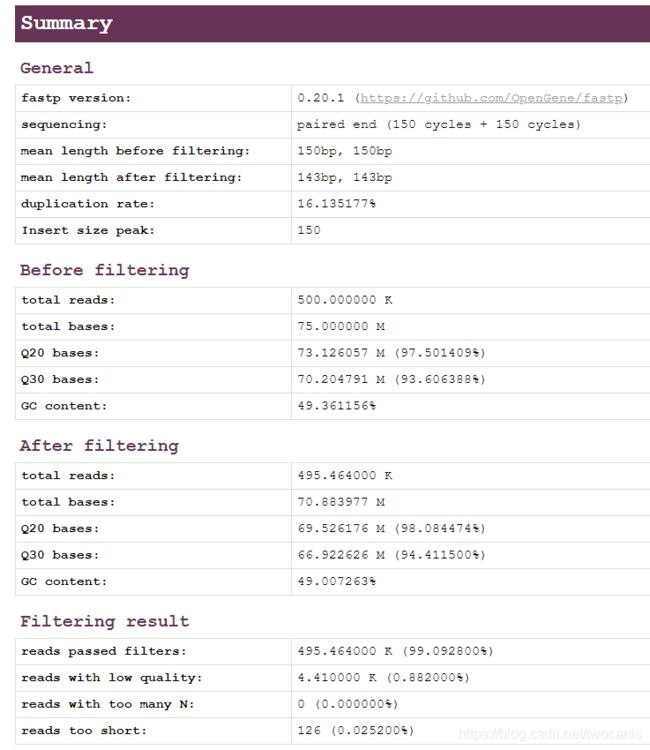
- General
版本号、序列循环数、质控之前的平均长度、质控之后的平均长度、插入片段的峰值 - Before filtering
数据质控之前的(反应测序质量):总的reads长度、总碱基长度、Q20合格率、Q30合格率、GC含量 - After filtering
质控之后的:内容同上 - Filtering result
reads的通过率、低质量的reads、含太多N值的reads
Adapter
即刚刚上面介绍的接头,这里两个文件(两端的reads)列出了从1到几十位的adapters的发生次数,以及其他未列出的接头数

Insert size estimation
配对末端重叠分析,不同长度的Insert在reads中占的比例,相当于是DNA被打断后的长度分布。当插入片段大小<30或> 270,或包含太多错误,则不能被read读取,比如我这里就有10.074194%的不可读reads)

Before filtering
质控之前的数据质量、碱基含量以及kmer分析等,可直接在网页上用鼠标拖动放大缩小以及查看具体数据细节,或进行图片保存等操作
-
reads质量
在不同位置上的碱基质量分布,一般来讲质量应 >30 且波动较小为不错的数据

-
碱基质量
read各个位置上碱基比例分布,这个是为了分析碱基的分离程度。何为碱基分离?已知AT配对,CG配对,假如测序过程是比较随机的话(随机意味着好),那么在每个位置上A和T比例应该差不多,C和G的比例也应该差不多,如上图所示,两者之间即使有偏差也不应该太大,最好平均在1%以内,如果过高,除非有合理的原因,比如某些特定的捕获测序所致,否则都需要注意是不是测序过程有什么偏差。

-
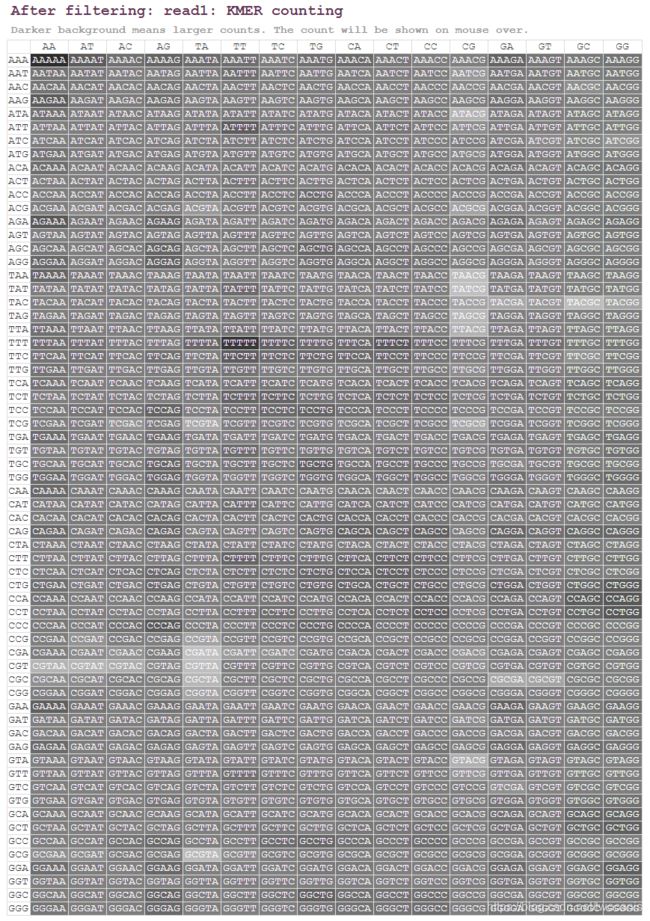
KMER计数
fastp对5个碱基长度的所有组合的出现次数进行了统计,然后把它放在了一张表格中,表格的每一个元素为深背景白字,背景越深,则表示重复次数越多。这样,一眼望去,就可以发现有哪些异常的信息。鼠标可停留在某一具体组合上看出现次数和平均占比。
剩下一部分After filtering就是质控之后结果,指标和质控之前一致,不赘述了。
以上就是刚接触fastp后做的一个学习笔记,基本上自己目前找到和理解的就这些,正在慢慢学习,欢迎一起讨论。
参考资料:
《全基因组测序WGS数据分析——3.数据质控》学习笔记
fastp:极速全能的FASTQ文件自动质控过滤校正预处理软件