Nat. Communications Biology2022 | PepNN+: 用于识别多肽结合位点的深度关注模型
论文标题:PepNN: a deep attention model for the identification of peptide binding sites
论文链接:PepNN: a deep attention model for the identification of peptide binding sites | Communications Biology
代码地址:oabdin / PepNN · GitLab
一、问题提出
基于结构和序列的方法来预测蛋白质上的肽结合位点。预测肽-蛋白相互作用的一个主要困难是肽的灵活性和它们在结合时发生构象变化的倾向。
在缺乏包括已解结构在内的充足实验数据的情况下,获得对肽-蛋白相互作用及其相关疾病状态的分子洞察力取决于计算模拟肽结合的能力。一种广泛使用的肽对接工具是FlexPepDock,通过从肽内的自由度取样来细化粗粒肽-蛋白质构象。但是肽对接方法往往不能准确识别天然复合物构象,受到多肽高度灵活性以及启发式评分固有误差的限制。机器学习方法为对接提供了潜在的替代方案,因为可以避免显式枚举构象空间的问题,并且可以直接从数据中学习评分指标。
一个重要障碍是缺乏可用多肽-蛋白的训练数据。为了克服这个问题,利用现有的蛋白质-蛋白质复合物信息,从而增加一个数量级的训练数据。
二、Methods
1、Datasets
分辨率至少为2.5 Å的晶体结构,包含超过30个氨基酸的链与25个或更少的氨基酸链的复合物,被认为是假定的肽蛋白复合物。使用FreeSASA,可以过滤出埋藏表面积(a buried surface area)小于400 Å2的复合物。利用MMseqs2以30%的识别阈值对剩余复合物中的受体序列进行聚类,并将聚类结果分别以90%和10%的比例分成训练集和验证集。在70%的覆盖率(MMseq2参数)下,与训练集或验证集序列的一致性超过30%(MMseq2参数)的测试集序列被删除。其余序列以90%同一性聚类,每个聚类只保留质心。结果数据集包含92个序列。
生成蛋白质片段-蛋白质复合物的数据集。使用Peptiderive Rosetta 程序, PDB扫描长度为5-25个氨基酸的蛋白质片段,当与另一条至少50个氨基酸的链配合时,具有高预测界面能。根据实际蛋白质-肽络合物数据集预测界面能的分布,过滤出络合物。
只有界面得分比肽-蛋白复合物分布的平均值高出小于一个标准差的复合物才得以维持。低于400 Å2的复合物再次被过滤掉。最终的数据集包含406,365个复合体。对于数据分割,复合体再次聚集在30%的同一性。
在这两个数据集中,结合残基被定义为蛋白质受体中与相互作用链中的重原子在6 Å以内的重原子残基。在测试集中70%的序列覆盖率下具有30%同一性的链被删除。
2、Input representation.
PepNN-Struct中,输入蛋白质结构使用图表示进行编码,同时在每个残基上添加额外的节点特征来编码侧链构象。在这种表示中,根据Cα与其他主原子的相对位置,在每个残基上定义一个局部坐标系。残基之间的边缘编码了有关残基之间距离的信息,从一个Cα到另一个Cα的相对方向,局部坐标系之间旋转矩阵的四元组表示,以及蛋白质序列中残基相对位置的嵌入。节点包括氨基酸identity 和the torsional backbone angles的one-hot。为了编码侧链构象的信息,计算每个残基上重侧链原子的质心。原子质心到Cα的方向用单位矢量表示,该单位矢量基于所定义的局部坐标系。距离使用径向基函数进行编码。
蛋白质和肽序列信息采用one-hot。使用预训练模型ProtBert将蛋白质序列embedding到PepNN-Seq中。
3、Model architecture.
PepNN与传统变压器的不同之处在于,而是利用reciprocal attention (类似于cross-attention)。
reciprocal attention modules:
Q、K来自当前类型数据信息(蛋白质/配体),V来自另一类型数据信息(配体/蛋白质)

利用随机搜索优化模型超参数, 总共进行了100次随机超参数试验
Training: Adam,对PepNN-Struct进行预训练和微调学习率为1e−4,对PepNN-Seq进行微调时的学习率为1e−5。加权交叉熵损失。此外,在微调过程中,用相同序列聚类中样本数的倒数对样本进行加权。在片段复合数据集和肽复合数据集的预训练步骤中,基于验证损失提前停止。在预训练阶段,训练最多有150,000次迭代,在微调阶段,训练最多35,000次迭代。
Scoring potential novel peptide binding sites.
通过向模型提供蛋白质序列/结构和长度为 10 的多甘氨酸序列作为肽,对人类蛋白质组和 PDB 中的蛋白质进行了肽不识别预测。以下公式用于为假定的肽结合位点分配分数:

p(r)为第i个残基的最高的结合概率,N为蛋白质残基总数,p为训练数据中结合位点分布的高斯拟合pdf, α为加权因子, 使用PepNN-Struct计算得分时,α设为0.955。使用Wilcoxon秩和检验对每个PFAM域与其余域的分布进行两两比较,并使用Benjamini-Hochberg程序进行多重检验校正。
三、Results
1、Transfer learning results in large improvements in model performance
通过两种方式使用迁移学习来提高模型性能。第一种是在一个较大的蛋白质片段-蛋白质复合物数据集上预训练模型,然后用较小的肽-蛋白质复合物数据集对模型进行微调。第二种是使用预训练语言模型ProtBert来embedding蛋白质序列。a表示使用预训练效果提升明显。b表示迁移ProtBert embedding给模型带来提升:
 |
 |
对PHF3的SPOC结构域的模型预测表明了这种性能上的差异,因为只有模型的预训练变体才能正确预测肽结合位点:

更一般地说,性能和绝对性能的改进都与预训练数据集中测试集示例与蛋白质的结构相似性无关:
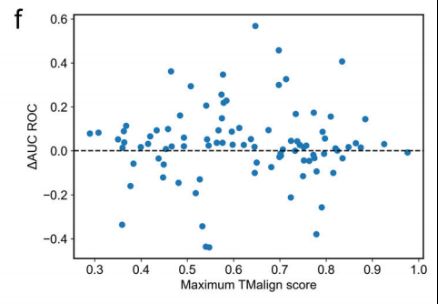
这表明,预训练有助于使参数更接近于一般肽结合位点预测的最佳值,而不是仅仅通过与片段复杂数据集中看到的模式匹配的示例来提高性能。
2、PepNN outperforms an equivalent Graph Transformer.
在一个独立的测试集TS092上评估模型,TS092是肽-蛋白复合物,与训练和预训练数据集没有冗余,以及先前研究的三个基准数据集。为对模型性能进行无偏估计,在评估测试集之前,在不同研究中使用的训练数据集上重新训练PepNN。毫不奇怪,发现PepNN-Struct始终优于PepNN-Seq:
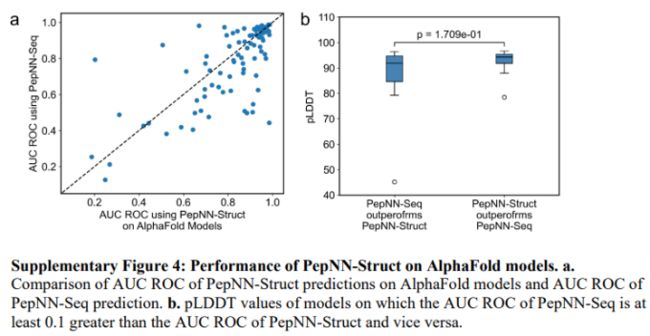
在大多数情况下,使用AlphaFold模型的基于结构的预测仍然比基于序列的预测更接近真实情况。为了评估这种趋势是否取决于AlphaFold生成的模型的质量,比较了PepNN-Struct表现更好的情况和PepNN-Struct表现更好的情况下,由AlphaFold生成的模型plddt的分布。虽然在PepNN-Seq表现较好的情况下,pLDDT平均较低(图4b),但差异在统计上并不显著(可能是因为AlphaFold在该测试数据集上生成了一致的高质量蛋白质模型):
3、Peptide-agnostic prediction allows the identification of putative novel peptide-binding proteins.
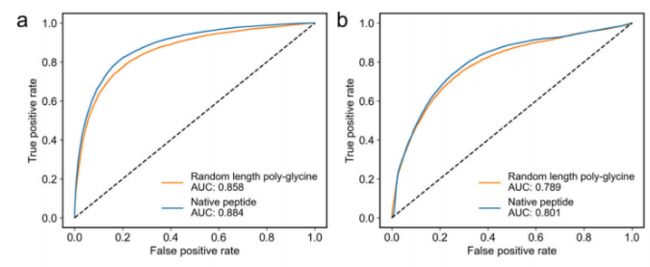
为了量化模型在进行预测时对蛋白质信息的依赖程度,测试了PepNN-Struct和PepNN-Seq使用随机长度的聚甘氨酸肽作为输入序列来预测肽结合位点的能力。虽然当给定天然肽序列时,模型的表现确实比给定聚甘氨酸序列时更好(DeLong检验中,PepNN-Struct和PepNN-Seq的p值都< 2.2e−16),但当给定聚甘氨酸肽时,ROC AUC的总体下降幅度很小:
比较模型分配给不同残基的概率表明,在PepNN-Struct的情况下,提供天然肽增加了模型在预测结合残基时的置信度。因此,提供天然肽序列对于减少假阴性非常重要:
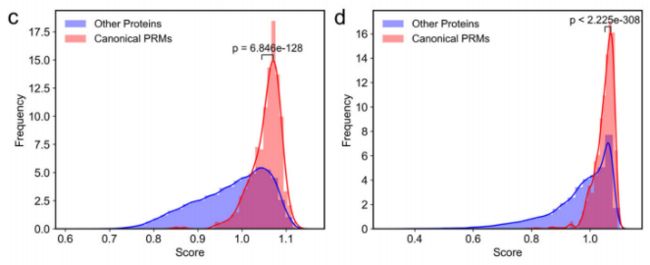
为评估模型区分肽结合模块和其他结构域的能力,比较了典型PRMs与其他蛋白质的得分分布。先前定义的模块化蛋白域s和肽结合域s被认为是典型的PRMs。在PDB和人类蛋白质组的情况下,典型PRMs的得分平均高于其他结构域:
4、Prediction of multiple peptide binding sites using PepNN.
在TS092中发现了两个这样的例子,其中一个蛋白质同时结合两个肽,当使用PepNN-Struct以肽不确定的方式预测这些蛋白质的结合位点时,两个结合位点都有一定的置信度预测,稍微偏向于一个。当使用天然肽序列进行预测时,对正确结合位点的置信度增加。在最初倾向于正确结合位点的情况下,对备选结合位点的置信度也会降低:

为了更广泛地评估PepNN-Struct预测多个结合位点的能力,对由多个PRMs组成的非冗余PDB链衍生的单个结构域和完整蛋白进行预测。
由于PepNN-Struct一致地预测了单个典型PRMs中的结合位点,单域和全蛋白预测的相关性表明预测了多个结合位点。对于大多数例子,对单个结构域和完整蛋白的预测具有良好的相关性。

唯一的例外是TCERG1蛋白的FF结构域,其中PepNN-Struct仅预测完整蛋白中FF结构域4和5的结合位点(图e):

有趣的是,先前的研究表明,当结合RNA-聚合酶c端结构域的肽时,只有结构域4和5中的残基参与相互作用(图f)。这支持了PepNNStruct将优先预测具有高肽结合倾向的位点的观点。