【无标题】
空间转录组自用笔记
- 前言
- 一、空间转录组分类
- 二、实验内容
-
- 1.选择二者之一
- 2.结合scRNA及核染色
- 3.空间转录组数据分析流程
-
- 3.1预处理
- 3.2用于下游分析的通用工具包
- 3.3空间特征识别
- 3.4去卷积
- 3.5插补和映射到单个细胞
- 3.6细胞间相互作用推断
前言
一、空间转录组分类
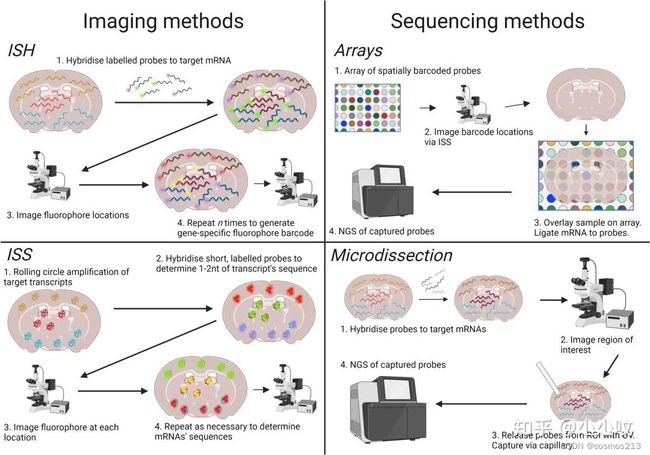
空间转录组技术主要分为两类:一类是基于杂交和成像的方法,另一类是基于测序的方法。
(一)基于成像的空间转录组学技术的基础:首先通过显微镜对mRNAs进行原位成像。当对mRNAs进行原位成像时,还必须有区分不同mRNA种类的方法,即原位杂交(ISH)或原位测序(ISS)。
(二)基于测序的空间转录组学技术的基础(测序是指NGS而不是ISS):首先是从组织中提取mRNAs,同时保留空间信息,随后通过高通量测序(NGS)技术对mRNA种类进行分析。保存空间信息的常见方法是:(1)通过直接捕获和记录位置,如通过显微切割和微流体技术;
(2)通过将mRNAs与芯片中的空间编码探针连接。此外,现在还有了进行空间基因组和蛋白质组实验的技术,比如DNA seqFISH+、DBiT-seq、t-CycIF和CODEX等。
————————————————
原文及图片来源:https://zhuanlan.zhihu.com/p/555201626
二、实验内容
1.选择二者之一
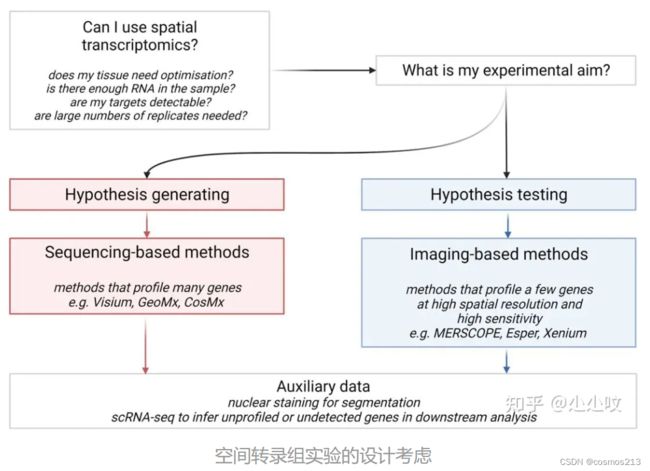
研究团队认为假设检验实验最适合ISH和ISS方法,如MERSCOPE、Esper和Xenium。
相反的无偏假设生成和图谱生成实验(可能具有较大的组织面积)最适合基于芯片的方法,如Visium和STomics。
————————————————
原文及图片来源:https://zhuanlan.zhihu.com/p/555201626
2.结合scRNA及核染色
许多空间技术并非检测的是真正的“单细胞”,其可以从同一组织的配套scRNA-seq数据集中获益。
对于靶向技术,无偏单细胞参考可用于推断未进行空间测量的基因表达或用于将单独成像的mRNA分配给单个细胞,已发表的多个生信工具可完成上述任务。
对于基于芯片的方法,通常使用单细胞参考来推断哪些细胞类型以及以何种比例导致每个捕获区域中的mRNA混合;这个过程称为去卷积。其次,在某些情况下,该参考可用于推断空间技术不能很好描述的基因的表达。总的来说,在设计空间转录组学实验时,建议在处理可分离的组织时考虑生成一个参考的scRNA-seq数据集,不过这对于像人脑这样的精细组织来说可能比较困难。
( ps: 无偏单细胞参考和单细胞参考的区别在于,无偏单细胞参考是指在单细胞测序中,对于每个细胞,都有一个相同的参考基因组,这样可以避免不同细胞之间的比较时引入偏差。而单细胞参考则是指对于每个细胞,都有一个不同的参考基因组,这样可以更好地反映不同细胞之间的差异性。
无偏单细胞参考和单细胞参考的实现方式是通过不同的算法来实现的。无偏单细胞参考可以通过将所有细胞的基因表达数据合并,然后进行标准化来实现。而单细胞参考则是通过将每个细胞的基因表达数据与一个参考基因组进行比对,然后进行标准化来实现。)
3.空间转录组数据分析流程
3.1预处理
基于ISH和ISS的典型方法将通过多轮杂交读取序列或基因特异性条形码,因此图像本身没有信息。将这些图像转换为基因点矩阵需要几个步骤:
(1)对图像进行滤波以去除背景和噪声。
(2)将来自不同杂交轮的图像对齐,以便每个杂交轮中相同的像素位置或斑点代表相同的转录本。
(3)每个点的信号被组合成一个条形码/序列,可以用于将点与基因匹配,与任何基因不匹配的信号被过滤。
(4)一个可选步骤是分割。用于预处理ISH或基于ISS的转录组学的工具通常针对特定的技术。对于Visium,10X Genomics已经发布了一个预处理管道–Space Ranger。
( 分割的目的是用亚细胞分辨率的空间数据来重建单细胞转录组。例如:
分割可用于基于成像的数据:通过从转录物物种推断并聚类图像的哪些区域可能包含一个细胞来重建单细胞转录组。也可用于基于亚细胞芯片的数据:分割将基因点矩阵转换为推断的基因-细胞矩阵。)
预处理数据的最后一步是对基因点矩阵进行统计转换,以考虑整个组织的mRNA捕获率的差异。这对通过所有技术产生的数据都是一个重要步骤,尤其是那些捕获率较低或不稳定的技术
3.2用于下游分析的通用工具包
对于空间数据有一系列不同的下游分析,具有不同的目标和不同的输入。
空间数据可以包括原始基因点矩阵、归一化矩阵或辅助数据,例如转录组分析之前拍摄的组织学图像的推断细胞类型和组织结构域。为了给这些数据提供统一的格式,并简化和标准化空间分析,开发了Giotto、Seurat、scanpy、STUtility、stLearn和squidpy等实用程序包。
他们的共同目标之一是,首先为空间数据矩阵和通过下游分析生成的相关辅助数据提供结构。后者可能包括降维(例如UMAP)、无偏聚类结果、注释和插补、映射或去卷积结果。第二,是提供完成所有这些过程的功能。第三,它们提供了数据可视化的功能,将空间转录组数据与叠加数据(如显微镜数据)相结合。最后,它们为质控、预处理和空间数据的专门分析技术提供了标准化的工作环境。
综上所述,建议对生信不是特别专业的生物医学研究人员使用Seurat和Scanpy,因为它们有大量的文档和庞大的用户群体,并建议寻求更专业的空间转录组学分析流程的研究人员使用Giotto、stLearn、STUtility和squidpy。
3.3空间特征识别
许多scRNA-seq分析的最初目标是确定细胞类型。空间转录组学分析的一个类似步骤可能是识别空间特征,如解剖学和微解剖学结构。为了识别结构,现有的算法可以将转录组相似的点或细胞以无偏的方式分组,以揭示基因表达的空间模式。然而,专门利用空间数据和识别组织域等特征的最新方法已经被开发出来,例如BayesSpace、XFuse、stLearn等。方法的选择将取决于可用的数据:对于基于芯片的低分辨率方法,BayesSpacem可能有利于提高分辨率,但如果有组织学图像可用,stLearn能够将它们集成到空间转录组数据的分析中。
或者,与其识别样本中细胞和斑点之间的异质性,不如直接搜索显示有偏见、非随机空间表达模式的基因。Giotto的空间可变基因选择方法在速度上优于一些旧方法,如SpatialDE、trendsceek和SPARK,鉴于空间转录组学的数据集不断增大的趋势,这是一个关键的问题。Sepal是一个较新的方法,它采取了一种新的方法,模拟单个物种的观察转录物在整个样本中扩散到随机分布所需的时间,这个指标推断了物种分布的空间结构程度。分组方法可用于提高速度,但这可能会导致空间细节的损失,这取决于所用分组的大小。
3.4去卷积
目前已发表的去卷积工具较多,包括SPOTlight、RCTD、cell2location、Tangram和destVI等,Seurat和Giotto等实用软件包也提供了去卷积方法。选择去卷积技术时,研究团队建议用户考虑运行时间,因为这一步骤可能需要大量的计算时间和功率。
href=“https://mp.weixin.qq.com/s?__biz=MzA4MDkwODYyOQ==&mid=2452653964&idx=1&sn=1c31a6743dc6696382eac4dffab62494&chksm=88593fb6bf2eb6a0f8096b3984ecc2650c0be19187d2bdb06ac991eeadf9d8a849d4949f833f&token=1805245652&lang=zh_CN&scene=21#wechat_redirect”> 10款空间转录组去卷积工具的综合比较
3.5插补和映射到单个细胞
一些计算方法旨在结合空间转录组和scRNA-seq,不是为了去卷积,而是为了推断未检测到的基因实际上可能在哪里表达。这允许用户“填补空白”,即空间转录组数据的目标性质,或者对于某些方法,低灵敏度意味着没有检测到基因,这项任务称为插补。相反,一些方法采用相反的方法,使用空间数据集推断scRNA-seq衍生的单细胞转录组的空间映射。
早期的整合方法是Seurat,它将单细胞转录组映射到空间坐标。最近的映射方法包括SpaOTsc,它依赖于优化的传输模型将单细胞转录组映射到空间数据,还包括推断配体-受体相互作用的功能。插补方法包括基于深度学习的gimVI;Tangram使用映射步骤通知插补过程;以及SpaGE,通过域适应来对齐空间和scRNA-seq数据,从而为插补提供信息。gimVI和Tangram是基于深度学习的方法,因此方法的选择可能取决于研究人员是否可以使用GPU计算资源。单细胞转录组的映射可能对非单细胞数据有用,例如未分割的基于成像的数据或来自任何基于测序的方法数据。相反,插补将有助于推断靶向空间转录组数据中的未测量基因。
3.6细胞间相互作用推断
分析转录组数据的一个共同目标是根据配体和受体的表达来推断细胞间的相互作用。通常情况下,细胞-细胞相互作用推断工具结合了被证明参与细胞间相互作用的蛋白质编码基因数据库,以及从基因表达数据推断相互作用概率的算法。已经开发了几种用于此目的的技术,包括SpaOTsc、cell2cell、MISTy和CellPhoneDB v.3.0,以及一种在通用空间转录组学分析软件包Giotto中实现的技术。
————————————————
原文及图片来源:https://zhuanlan.zhihu.com/p/555201626