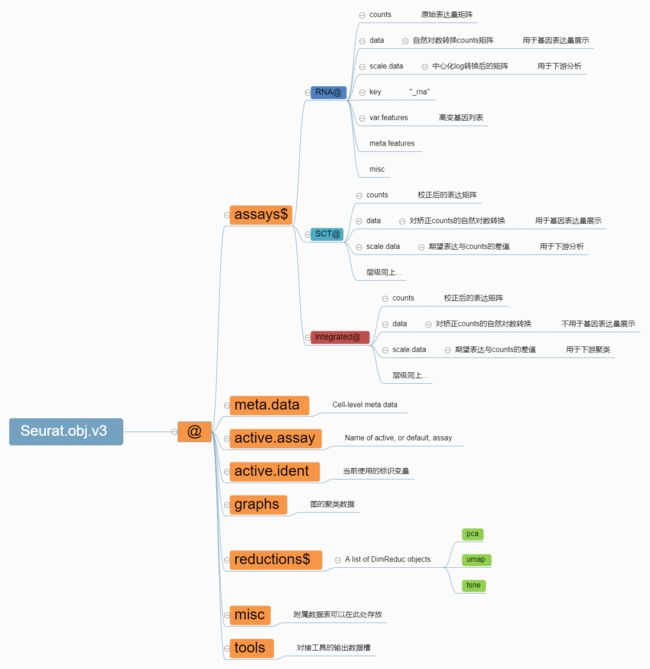
- Ollama平台里最流行的embedding模型: nomic-embed-text 模型介绍和实践
skywalk8163
人工智能embedding人工智能服务器
nomic-embed-text模型介绍nomic-embed-text是一个基于SentenceTransformers库的句子嵌入模型,专门用于特征提取和句子相似度计算。该模型在多个任务上表现出色,特别是在分类、检索和聚类任务中。其核心优势在于能够生成高质量的句子嵌入,这些嵌入在语义上非常接近,从而在相似度计算和分类任务中表现优异。之所以选用这个模型,是因为在Ollama网站查找这个模型,发现
- 【数据分析】多数据集网络分析:探索健康与退休研究中的变量关系
生信学习者1
数据分析(2025版)数据分析r语言数据挖掘数据可视化
禁止商业或二改转载,仅供自学使用,侵权必究,如需截取部分内容请后台联系作者!文章目录介绍加载R包数据下载导入数据数据预处理函数网络分析画图保存图片总结系统信息介绍在医学和社会科学研究中,理解多个变量之间的复杂关系对于揭示潜在的病理生理机制和社会行为模式至关重要。本文介绍了一种基于R语言的网络分析方法,用于探索HRS(健康与退休研究)及其类似研究(CHARLS、ELSA、MHAS、SHARE)中的变
- R语言舆情监控与可视化统计
q56731523
r语言开发语言爬虫
用R语言进行舆情监控并且做到可视化,对我来说,总体难度还算可以,主要是舆情监控通常涉及文本数据的收集(如社交媒体、新闻评论),然后进行情感分析,最后通过图表展示结果。步骤看似简单实则一点也不简单。以下就是我使用R语言进行舆情监控和可视化统计的完整示例。该方案包括文本情感分析和时间趋势可视化:#加载必要的包library(tidyverse)#数据处理和可视化library(tidytext)#文本
- R 语言数据框连接操作详解:join 与 merge 方法对比
晚风keeper
r语言开发语言学习笔记学习方法
在数据分析工作中,我们经常需要将多个数据集按照某些条件进行合并。R语言提供了多种数据框连接方法,本文将详细介绍如何使用dplyr包的join系列函数和基础R的merge函数进行数据框的各种连接操作,并对比它们之间的差异。一、数据框连接操作概述数据框连接是将两个或多个数据框按照某些共同的列或条件组合成一个新的数据框的过程。常见的连接类型包括:左连接(LeftJoin):保留左数据框的所有行,匹配右数
- Readr 项目安装与配置指南
芮奕滢Kirby
Readr项目安装与配置指南readr项目地址:https://gitcode.com/gh_mirrors/rea/readr1.项目基础介绍readr是一个R语言的开源项目,由HadleyWickham创建和维护。该项目的主要目的是提供一种快速且友好的方式来读取分隔文件(如CSV和TSV)中的矩形数据。readr能够解析多种数据类型,并在解析过程中提供详细的错误报告,以便用户能够快速识别和解决
- 4篇2章5节:ANOVA 功效的单次精确模拟与可视化全解析
MD分析
用R探索医药数据科学r语言-4.2.1r语言功效曲线单次精确模拟分析
在医学研究尤其是糖尿病等干预性试验中,精准的实验设计与功效分析是确保研究价值的关键。R语言为重复测量方差分析(ANOVA)提供了强大工具,从实验设计构建、单次精确模拟分析,到功效曲线可视化,覆盖研究全流程。本文结合糖尿病胰岛素治疗试验案例,深度拆解函数的应用逻辑,手把手教你用数据驱动实验设计,让“样本量规划”“效应检测能力”从抽象概念变为可操作、可视化的研究支撑。一、相关函数的介绍在医学研究中,实
- R语言如何接入实时行情接口
目录1.安装必要的R包2.导入库3.连接WebSocket4.处理连接成功后的操作5.处理接收到的消息6.处理连接关闭和错误7.发送心跳数据8.自动重连机制9.启动连接和重连总结在数据分析和金融研究中,实时行情数据的获取至关重要,但市面上的实时行情接口并不多,本文将一步步教你如何使用R语言接入实时行情接口,获取来自WebSocket的实时数据。1.安装必要的R包首先,确保你已安装了以下R包,用于处
- 【R语言】Can‘t subset elements that don‘t exist.
新子y
r语言开发语言excel
Errorin`select()`:ℹInargument:`all_of(label_col)`.Causedbyerrorin`all_of()`:!Can'tsubsetelementsthatdon'texist.✖Element`Label`doesn'texist.Run`rlang::last_trace()`toseewheretheerroroccurred.原文中文解释涉及关键
- r读取文件夹下的所有csv文件_R语言读取文件夹下多个文件并进行合并数据生成总数据文件...
seiji morisako
r读取文件夹下的所有csv文件
在流水化办公中,通常有格式统一的表格文件产生,但是到最后要将这一堆表格文件整合为大表却很揪心,累断手,如何用R语言进行一次性导入整合呢?假设我们将D:/input文件夹作为需要导入的表格的存放点,文件夹内所有文件均为此次需要导入数据,那么可以用以下程序进行操作setwd("D:/")#设定工作目录为D盘a=list.files("input")#list.files命令将input文件夹下所有文件
- 从0开始学习R语言--Day41--Moran‘s I
Chef_Chen
学习
在处理带有空间特征的数据,我们往往都直接一股脑地处理数据点,但很多时候,空间上的信息对于处理后续衍生出来的问题会有很大帮助,例如对于城市里大小县城的发展情况,只知道单一县城的经济发展曲线,很难解释一些拐点和突然的攀升,而如果知道相邻县城存在经济发展飞快的例子,可能就是被带动了经济水平;亦或者是在处理社交网络的好有问题时,只知道谁和谁是朋友(类似于空间矩阵),是无法推断出经济收入相似的推论的,所以说
- 结构方程模型(SEM)高阶应用系列
梦想的初衷~
结构方程生态环境python开发语言结构方程
结构方程模型(StructuralEquationModeling)是分析多变量间因果关系的利器,在众多学科领域具有巨大应用潜力。我们前期推出的《基于R语言结构方程模型》通过结构方程原理介绍、结构方程全局和局域估计、模型构建和调整、潜变量分析、复合变量分析及结构方程贝叶斯方法实现等一系列专题的介绍及大量案例讲解,由浅入深地系统介绍了结构方程模型的建立、拟合、评估、筛选和结果展示全过程,得到学员广泛
- r语言改变数据框列名_数据决定离线强化学习将如何改变我们的语言习惯
杨_明
python大数据人工智能java机器学习
r语言改变数据框列名重点(Tophighlight)Aridesharingcompanycollectsadatasetofpricinganddiscountdecisionswithcorrespondingchangesincustomeranddriverbehavior,inordertooptimizeadynamicpricingstrategy.Anonlinevendorrec
- 【数据分析】R语言基于虚弱指数的心血管疾病风险评估
生信学习者1
数据分析(2025版)数据分析r语言数据挖掘数据可视化
禁止商业或二改转载,仅供自学使用,侵权必究,如需截取部分内容请后台联系作者!文章目录介绍加载R包数据下载导入数据数据预处理画图其他1其他2其他3其他4总结系统信息介绍生存分析是医学和生物统计学中常用的方法,用于研究事件(如疾病发生、死亡等)发生的时间和相关影响因素。本文介绍了一种基于R语言的生存分析方法,用于评估虚弱指数(FrailtyIndex,FI)对心血管疾病(CVD)发生风险的影响。通过这
- 【科研绘图系列】R语言绘制论文组合图(multiple plots)
生信学习者1
SCI科研绘图系列(2025版)r语言数据分析数据挖掘数据可视化
禁止商业或二改转载,仅供自学使用,侵权必究,如需截取部分内容请后台联系作者!文章目录介绍加载R包数据下载函数数据预处理画图1画图2画图3画图4画图5画图6总结系统信息介绍这段代码是一个用于生成多种复杂数据可视化的R脚本,主要利用ggplot2、tidyverse和自定义函数来处理和展示与小鼠实验相关的数据。它通过读取、处理数据,并生成多种图形,旨在清晰地展示不同实验组的小鼠在不同时间点的抗体浓度和
- LSA主题模型:基于奇异值分解的主题模型
AI天才研究院
AI人工智能与大数据AI大模型企业级应用开发实战计算计算科学神经计算深度学习神经网络大数据人工智能大型语言模型AIAGILLMJavaPython架构设计AgentRPA
LSA主题模型:基于奇异值分解的主题模型1.背景介绍主题模型是一种无监督的机器学习技术,用于发现大规模文本语料库中隐藏的语义结构。它能够自动识别文档集合中的主题,并根据这些主题对文档进行聚类和分类。主题模型在文本挖掘、信息检索、推荐系统等领域有着广泛的应用。LSA(LatentSemanticAnalysis)是一种经典的主题模型算法,基于奇异值分解(SVD)对词-文档矩阵进行分解,从而揭示词语和
- 【机器学习笔记 Ⅱ】9 模型评估
巴伦是只猫
机器学习机器学习笔记人工智能
评估机器学习模型是确保其在实际应用中有效性和可靠性的关键步骤。以下是系统化的评估方法,涵盖分类、回归、聚类等任务的评估指标和技术:一、分类模型评估1.基础指标2.高级指标ROC-AUC:通过绘制真正例率(TPR)vs假正例率(FPR)曲线下面积评估模型整体性能。AUC=1:完美分类;AUC=0.5:随机猜测。适用于二分类及多分类(OvR或OvO策略)。混淆矩阵:可视化模型在各类别上的具体错误(如将
- 如何自定义R语言函数?参数中的省略号`...`有什么用?
「已注销」
python编程语言java人工智能c++
学习R未必要学习很多工具包,有时候根据自己的理解去自定义函数也是一个不错的选择。本篇推文主要介绍两方面的内容:在R语言中自定义函数的一般方法;函数参数中...的作用。在看函数的帮助文档时会发现许多函数的参数中都有...符号,它是表示被省略的参数吗?如果是,作者为什么会省略它?如果不是,那又表示什么含义呢?不久前,学堂君分享了自己编写的计算空间可达性的函数,详见推文:两步移动搜索法(2SFCA)计算
- Logistic回归预测模型2:R语言实现模型的内部和外部验证
前面我们讲了logistic回归预测模型的建立,今天介绍的是模型的验证,可以在训练集和验证集中通过ROC曲线、校准曲线和决策曲线分别进行验证。1、原始数据原始数据分为训练集和验证集,其中训练集用于模型的构建和内部验证,验证集用于外部验证。两个数据集都包含5列,且列名相同。组别Group为因变量,1代表阳性结局,0代表阴性结局。自变量1和4为连续性变量,自变量2和3为二分类变量。2、安装所需要的R包
- R 列表:深入解析与高效应用
沐知全栈开发
开发语言
R列表:深入解析与高效应用引言在R语言中,列表(List)是一种非常重要的数据结构,它允许我们将不同类型的数据组合在一起。列表在数据分析和统计建模中扮演着至关重要的角色。本文将深入探讨R列表的概念、创建方法、操作技巧以及在实际应用中的高效使用。R列表概述定义R列表是一种可以包含多种数据类型的数据结构,如数值、字符、逻辑值、其他列表等。列表可以看作是一个容器,可以存储任意数量的元素。类型R列表分为两
- 【机器学习笔记 Ⅱ】10 完整周期
机器学习的完整生命周期(End-to-EndPipeline)机器学习的完整周期涵盖从问题定义到模型部署的全过程,以下是系统化的步骤分解和关键要点:1.问题定义(ProblemDefinition)目标:明确业务需求与机器学习任务的匹配性。关键问题:这是分类、回归、聚类还是强化学习问题?成功的标准是什么?(如准确率>90%、降低10%成本)输出:项目目标文档(含评估指标)。2.数据收集(DataC
- 机器学习宝典——第6章
爱看烟花的码农
机器学习人工智能
第6章:聚类算法(Clustering)你好,同学!欢迎来到无监督学习的世界。与监督学习不同,这里的我们没有“标准答案”(标签),我们的目标是在数据中发现隐藏的、内在的结构。聚类算法就是实现这一目标的核心工具,它试图将数据集中的样本划分为若干个不相交的子集,我们称之为“簇”(cluster)。本章我们将深入探讨三种最具代表性的聚类算法:K-均值(K-Means)、层次聚类(Hierarchical
- R 语言安装使用教程
小奇JAVA面试
安装使用教程r语言开发语言
一、R语言简介R是一种用于统计分析、数据挖掘和可视化的编程语言和环境。它在学术界和数据分析领域中广泛使用,拥有丰富的统计函数库和绘图功能。二、安装R语言2.1下载R安装包前往CRAN官网下载适合你操作系统的安装程序:官网地址:https://cran.r-project.org/2.2Windows安装下载.exe安装包;双击安装程序,按默认选项一路安装即可;安装完成后,可通过RGUI或命令行启动
- R语言学习笔记—删除对象
w1149033842
R语言
1.删除环境中的对象Arm(A)2.删除环境中的所有对象rm(list=is())3.删除除了A和B以外的所有对象allobj<-is()rm(list=allobj[which(allobj!="A"&allobj!="B")])
- R语言的游戏开发
柳婉晴
包罗万象golang开发语言后端
R语言在游戏开发中的应用随着科技的发展,游戏行业已经成为一个巨大的市场。虽然通常我们会认为游戏开发主要是使用C++、C#、JavaScript等语言,但实际上,R语言在游戏开发中也有其独特的应用,尤其是在数据分析和可视化方面。本文将探讨R语言在游戏开发中的应用,涵盖它的基础、游戏设计的复杂性、实际案例分析、以及未来的发展方向。一、R语言基础R语言是一种用于统计计算和数据分析的编程语言。它具有强大的
- R语言的软件开发工具
纪霁然
包罗万象golang开发语言后端
R语言的软件开发工具引言R语言因其强大的数据分析能力和丰富的统计包,自发布以来便广受欢迎。随着数据科学和分析的迅猛发展,R语言也逐渐成为数据分析、机器学习和统计建模领域的重要工具。为了更好地利用R语言进行软件开发,许多软件开发工具和环境应运而生。本文将深入探讨R语言的主要开发工具,帮助开发者更高效地进行数据处理和分析。1.R和RStudio基础R语言本身是一个用于统计计算和图形绘制的编程语言,而R
- R语言初学者爬虫简单模板
q56731523
r语言爬虫开发语言iphone
习惯使用python做爬虫的,反过来使用R语言可能有点不太习惯,正常来说R语言好不好学完全取决于你的学习背景以及任务复杂情况。对于入门学者来说,R语言使用rvest+httr组合,几行代码就能完成简单爬取(比Python的Scrapy简单得多),R语言数据处理优势明显,爬取后可直接用dplyr/tidyr清洗,小打小闹用R语言完全没问题,如果是企业级大型项目还是有限考虑python,综合成本还是p
- R语言开发记录,一
[email protected]
R语言r语言开发语言
1.清理环境rm(list=ls())gc()rm(list=ls())作用:删除当前R工作环境中所有的对象(变量、函数、数据框等)。解释:ls():列出当前环境中所有对象的名字。list=ls():将这些名字作为一个列表传给rm()函数。rm():移除这些对象。效果:相当于“清空内存”,让工作空间恢复到干净状态。gc()作用:手动触发垃圾回收(garbagecollection)。效果:释放R不
- 从零到精通:Linux上的Conda环境详细教程
第一章:Conda简介Conda的定义Conda是一个开源的包管理系统和环境管理系统,可以在多个平台上安装、运行和更新软件包和依赖项。Conda最初是为Python和R语言的数据科学包创建的,但现在支持多种编程语言和工具。Conda的主要功能和优势包管理:Conda能够自动处理包的依赖关系,确保每个包所需的库和工具都被正确安装。它支持从各种渠道安装包,如CondaForge和Anaconda官方仓
- 【2025CVPR】SEC-Prompt:少样本增量学习中的语义互补提示模型详解
清风AI
生成对抗网络人工智能神经网络pcm目标跟踪深度学习计算机视觉
目录一、研究背景:少样本增量学习的挑战二、SEC-Prompt核心原理1.自适应层次化查询(AdaptiveHierarchicalQuery)2.语义互补提示机制(1)判别性提示(D-Prompt)(2)非判别性提示(ND-Prompt)3.训练策略创新(1)判别性提示聚类损失(2)ND-Prompt数据增强三、模型架构图解四、关键创新点五、实验结果对比1.ImageNet-R结果2.CUB20
- R语言 绘制上下双向分布柱状图
话不多说,直接上干货library(ggplot2)library(tidyr)set.seed(123)#设置随机种子保证可重现df<-data.frame(Type=rep(letters[1:5],each=5),Sample=paste("sample",rep(1:5,times=5),sep=""),Up=round(runif(25,min=0,max=100),1),Down=ro
- ASM系列四 利用Method 组件动态注入方法逻辑
lijingyao8206
字节码技术jvmAOP动态代理ASM
这篇继续结合例子来深入了解下Method组件动态变更方法字节码的实现。通过前面一篇,知道ClassVisitor 的visitMethod()方法可以返回一个MethodVisitor的实例。那么我们也基本可以知道,同ClassVisitor改变类成员一样,MethodVIsistor如果需要改变方法成员,注入逻辑,也可以
- java编程思想 --内部类
百合不是茶
java内部类匿名内部类
内部类;了解外部类 并能与之通信 内部类写出来的代码更加整洁与优雅
1,内部类的创建 内部类是创建在类中的
package com.wj.InsideClass;
/*
* 内部类的创建
*/
public class CreateInsideClass {
public CreateInsideClass(
- web.xml报错
crabdave
web.xml
web.xml报错
The content of element type "web-app" must match "(icon?,display-
name?,description?,distributable?,context-param*,filter*,filter-mapping*,listener*,servlet*,s
- 泛型类的自定义
麦田的设计者
javaandroid泛型
为什么要定义泛型类,当类中要操作的引用数据类型不确定的时候。
采用泛型类,完成扩展。
例如有一个学生类
Student{
Student(){
System.out.println("I'm a student.....");
}
}
有一个老师类
- CSS清除浮动的4中方法
IT独行者
JavaScriptUIcss
清除浮动这个问题,做前端的应该再熟悉不过了,咱是个新人,所以还是记个笔记,做个积累,努力学习向大神靠近。CSS清除浮动的方法网上一搜,大概有N多种,用过几种,说下个人感受。
1、结尾处加空div标签 clear:both 1 2 3 4
.div
1
{
background
:
#000080
;
border
:
1px
s
- Cygwin使用windows的jdk 配置方法
_wy_
jdkwindowscygwin
1.[vim /etc/profile]
JAVA_HOME="/cgydrive/d/Java/jdk1.6.0_43" (windows下jdk路径为D:\Java\jdk1.6.0_43)
PATH="$JAVA_HOME/bin:${PATH}"
CLAS
- linux下安装maven
无量
mavenlinux安装
Linux下安装maven(转) 1.首先到Maven官网
下载安装文件,目前最新版本为3.0.3,下载文件为
apache-maven-3.0.3-bin.tar.gz,下载可以使用wget命令;
2.进入下载文件夹,找到下载的文件,运行如下命令解压
tar -xvf apache-maven-2.2.1-bin.tar.gz
解压后的文件夹
- tomcat的https 配置,syslog-ng配置
aichenglong
tomcathttp跳转到httpssyslong-ng配置syslog配置
1) tomcat配置https,以及http自动跳转到https的配置
1)TOMCAT_HOME目录下生成密钥(keytool是jdk中的命令)
keytool -genkey -alias tomcat -keyalg RSA -keypass changeit -storepass changeit
- 关于领号活动总结
alafqq
活动
关于某彩票活动的总结
具体需求,每个用户进活动页面,领取一个号码,1000中的一个;
活动要求
1,随机性,一定要有随机性;
2,最少中奖概率,如果注数为3200注,则最多中4注
3,效率问题,(不能每个人来都产生一个随机数,这样效率不高);
4,支持断电(仍然从下一个开始),重启服务;(存数据库有点大材小用,因此不能存放在数据库)
解决方案
1,事先产生随机数1000个,并打
- java数据结构 冒泡排序的遍历与排序
百合不是茶
java
java的冒泡排序是一种简单的排序规则
冒泡排序的原理:
比较两个相邻的数,首先将最大的排在第一个,第二次比较第二个 ,此后一样;
针对所有的元素重复以上的步骤,除了最后一个
例题;将int array[]
- JS检查输入框输入的是否是数字的一种校验方法
bijian1013
js
如下是JS检查输入框输入的是否是数字的一种校验方法:
<form method=post target="_blank">
数字:<input type="text" name=num onkeypress="checkNum(this.form)"><br>
</form>
- Test注解的两个属性:expected和timeout
bijian1013
javaJUnitexpectedtimeout
JUnit4:Test文档中的解释:
The Test annotation supports two optional parameters.
The first, expected, declares that a test method should throw an exception.
If it doesn't throw an exception or if it
- [Gson二]继承关系的POJO的反序列化
bit1129
POJO
父类
package inheritance.test2;
import java.util.Map;
public class Model {
private String field1;
private String field2;
private Map<String, String> infoMap
- 【Spark八十四】Spark零碎知识点记录
bit1129
spark
1. ShuffleMapTask的shuffle数据在什么地方记录到MapOutputTracker中的
ShuffleMapTask的runTask方法负责写数据到shuffle map文件中。当任务执行完成成功,DAGScheduler会收到通知,在DAGScheduler的handleTaskCompletion方法中完成记录到MapOutputTracker中
- WAS各种脚本作用大全
ronin47
WAS 脚本
http://www.ibm.com/developerworks/cn/websphere/library/samples/SampleScripts.html
无意中,在WAS官网上发现的各种脚本作用,感觉很有作用,先与各位分享一下
获取下载
这些示例 jacl 和 Jython 脚本可用于在 WebSphere Application Server 的不同版本中自
- java-12.求 1+2+3+..n不能使用乘除法、 for 、 while 、 if 、 else 、 switch 、 case 等关键字以及条件判断语句
bylijinnan
switch
借鉴网上的思路,用java实现:
public class NoIfWhile {
/**
* @param args
*
* find x=1+2+3+....n
*/
public static void main(String[] args) {
int n=10;
int re=find(n);
System.o
- Netty源码学习-ObjectEncoder和ObjectDecoder
bylijinnan
javanetty
Netty中传递对象的思路很直观:
Netty中数据的传递是基于ChannelBuffer(也就是byte[]);
那把对象序列化为字节流,就可以在Netty中传递对象了
相应的从ChannelBuffer恢复对象,就是反序列化的过程
Netty已经封装好ObjectEncoder和ObjectDecoder
先看ObjectEncoder
ObjectEncoder是往外发送
- spring 定时任务中cronExpression表达式含义
chicony
cronExpression
一个cron表达式有6个必选的元素和一个可选的元素,各个元素之间是以空格分隔的,从左至右,这些元素的含义如下表所示:
代表含义 是否必须 允许的取值范围 &nb
- Nutz配置Jndi
ctrain
JNDI
1、使用JNDI获取指定资源:
var ioc = {
dao : {
type :"org.nutz.dao.impl.NutDao",
args : [ {jndi :"jdbc/dataSource"} ]
}
}
以上方法,仅需要在容器中配置好数据源,注入到NutDao即可.
- 解决 /bin/sh^M: bad interpreter: No such file or directory
daizj
shell
在Linux中执行.sh脚本,异常/bin/sh^M: bad interpreter: No such file or directory。
分析:这是不同系统编码格式引起的:在windows系统中编辑的.sh文件可能有不可见字符,所以在Linux系统下执行会报以上异常信息。
解决:
1)在windows下转换:
利用一些编辑器如UltraEdit或EditPlus等工具
- [转]for 循环为何可恨?
dcj3sjt126com
程序员读书
Java的闭包(Closure)特征最近成为了一个热门话题。 一些精英正在起草一份议案,要在Java将来的版本中加入闭包特征。 然而,提议中的闭包语法以及语言上的这种扩充受到了众多Java程序员的猛烈抨击。
不久前,出版过数十本编程书籍的大作家Elliotte Rusty Harold发表了对Java中闭包的价值的质疑。 尤其是他问道“for 循环为何可恨?”[http://ju
- Android实用小技巧
dcj3sjt126com
android
1、去掉所有Activity界面的标题栏
修改AndroidManifest.xml 在application 标签中添加android:theme="@android:style/Theme.NoTitleBar"
2、去掉所有Activity界面的TitleBar 和StatusBar
修改AndroidManifes
- Oracle 复习笔记之序列
eksliang
Oracle 序列sequenceOracle sequence
转载请出自出处:http://eksliang.iteye.com/blog/2098859
1.序列的作用
序列是用于生成唯一、连续序号的对象
一般用序列来充当数据库表的主键值
2.创建序列语法如下:
create sequence s_emp
start with 1 --开始值
increment by 1 --増长值
maxval
- 有“品”的程序员
gongmeitao
工作
完美程序员的10种品质
完美程序员的每种品质都有一个范围,这个范围取决于具体的问题和背景。没有能解决所有问题的
完美程序员(至少在我们这个星球上),并且对于特定问题,完美程序员应该具有以下品质:
1. 才智非凡- 能够理解问题、能够用清晰可读的代码翻译并表达想法、善于分析并且逻辑思维能力强
(范围:用简单方式解决复杂问题)
- 使用KeleyiSQLHelper类进行分页查询
hvt
sql.netC#asp.nethovertree
本文适用于sql server单主键表或者视图进行分页查询,支持多字段排序。KeleyiSQLHelper类的最新代码请到http://hovertree.codeplex.com/SourceControl/latest下载整个解决方案源代码查看。或者直接在线查看类的代码:http://hovertree.codeplex.com/SourceControl/latest#HoverTree.D
- SVG 教程 (三)圆形,椭圆,直线
天梯梦
svg
SVG <circle> SVG 圆形 - <circle>
<circle> 标签可用来创建一个圆:
下面是SVG代码:
<svg xmlns="http://www.w3.org/2000/svg" version="1.1">
<circle cx="100" c
- 链表栈
luyulong
java数据结构
public class Node {
private Object object;
private Node next;
public Node() {
this.next = null;
this.object = null;
}
public Object getObject() {
return object;
}
public
- 基础数据结构和算法十:2-3 search tree
sunwinner
Algorithm2-3 search tree
Binary search tree works well for a wide variety of applications, but they have poor worst-case performance. Now we introduce a type of binary search tree where costs are guaranteed to be loga
- spring配置定时任务
stunizhengjia
springtimer
最近因工作的需要,用到了spring的定时任务的功能,觉得spring还是很智能化的,只需要配置一下配置文件就可以了,在此记录一下,以便以后用到:
//------------------------定时任务调用的方法------------------------------
/**
* 存储过程定时器
*/
publi
- ITeye 8月技术图书有奖试读获奖名单公布
ITeye管理员
活动
ITeye携手博文视点举办的8月技术图书有奖试读活动已圆满结束,非常感谢广大用户对本次活动的关注与参与。
8月试读活动回顾:
http://webmaster.iteye.com/blog/2102830
本次技术图书试读活动的优秀奖获奖名单及相应作品如下(优秀文章有很多,但名额有限,没获奖并不代表不优秀):
《跨终端Web》
gleams:http